
Abstract
Background
Enterococcus faecalis is a ubiquitous member of the gut microbiota and has emerged as a life- threatening multidrug-resistant (MDR) nosocomial pathogen. The aim of this study was to survey the prevalence of multidrug-resistant and epidemiologically important strains of E. faecalis in the western region of Saudi Arabia using phenotypic and whole genome sequencing approaches.
Methods
In total, 155 patients positive for E. faecalis infection were included in this study. The isolates were identified by MALDI-TOF, and screen for antimicrobial resistance using VITEK-2 system. Genome sequencing was performed with paired-end strategy using MiSeq platform.
Results
Seventeen sequence types (STs) were identified through multilocus sequence typing (MLST) analysis of the E. faecalis genomes, including two novels STs (ST862 and ST863). The most common STs in the Saudi patients were ST179 and ST16 from clonal complex 16 (CC16). Around 96% (n = 149) isolates were MDR. The antibiotics quinupristin/dalfopristin, clindamycin, and erythromycin demonstrated almost no coverage, and high-level streptomycin, gentamycin, and ciprofloxacin demonstrated suboptimal coverage. Low resistance was observed against vancomycin, linezolid, and ampicillin. Moreover, 34 antimicrobial resistance genes and variants, and three families of insertion sequences were found in the E. faecalis genomes, which likely contributed to the observed antimicrobial resistance. Twenty-two virulence factors, which were mainly associated with biofilm formation, endocarditis, cell adherence, and colonization, were detected in the isolates.
Conclusions
Diverse STs of E. faecalis, including strains associated with common nosocomial infections are circulating in the healthcare facility of Saudi Arabia and carried multi-drug resistance, which has important implications for infection control.
Similar content being viewed by others

Background
Over the past decades, enterococci have emerged as the most prevalent nosocomial pathogens. Once viewed as part of the normal gut flora with little clinical significance, and they are now recognized as the cause of several types of community- and hospital-acquired infections, including life-threating bloodstream infections, endocarditis, meningitis, and urinary tract infections [1]. Disease outbreaks from antimicrobial-resistant enterococci have occurred in the United States and Europe [2, 3], and the European Centre for Disease Prevention and Control (ECDC) has reported that enterococci are the most commonly isolated bacteria after Escherichia coli and Staphylococcus aureus from healthcare-associated infections in Europe [4]. Furthermore, nosocomial infections caused by vancomycin-resistant enterococci (VRE) represent a serious clinical problem in healthcare facilities in many countries worldwide [5, 6].
Enterococcus faecalis is the most common enterococcal species associated with nosocomial infections, accounting for 80–90% of the infections, followed by Enterococcus faecium (5–10%) [7]. Other species of enterococci rarely cause infection [2]. Genotyping of E. faecalis and E. faecium by multilocus sequence typing (MLST) and other methods revealed that distinct clones of these species are associated with hospital infections/outbreaks; these clones are referred to as high-risk enterococcal clonal complexes [8, 9]. In recent years, these species have shown increasing resistance to several antibiotics, including penicillin, aminoglycosides, and glycopeptides, which thus limits antimicrobial therapeutic options [10]. Glycopeptide resistance may be due to the acquisition of van genes, whereas vanC1 and vanC2/3 are responsible for intrinsic resistance in enterococci [11]. Vancomycin resistance that mainly arises from the vanA gene cluster is commonly identified on the mobile genetic element Tn1546 [12]. The mobile genetic elements from enterococci have also recently been shown to be able to transfer vancomycin resistance to more pathogenic bacteria such as S. aureus [13]. Monitoring the antimicrobial resistance (AMR) in enterococci from clinical specimens is essential for controlling the spread of resistance genes against vancomycin and other antibiotics.
Available literature has highlighted an increasing prevalence of multi-drug resistant (MDR) E. faecalis in the eastern region of Saudi Arabia [14]. However, no molecular characterization of MDR E. faecalis isolates from Saudi Arabia, and neighboring countries has been performed. An active molecular epidemiology program is critical for creating basic knowledge about local microorganisms and their resistance to refine policies on controlling infections from antimicrobial-resistant bacteria in hospitals and other healthcare facilities within the country. The main objective of this study was to perform antibiotic susceptibility, and genomic analysis of clinical E. faecalis isolates from the western part of the country. The isolates were evaluated for the presence of virulence and antimicrobial resistance genes (ARGs). MLST analysis was performed to track the global distribution of the E. faecalis sequence types (STs) identified in this study.
Materials and methods
Samples collection
King Abdulaziz University Hospital (KAUH) is an 845-bed teaching hospital that mainly serves the western region of Saudi Arabia. The E. faecalis strains isolated from patients at KAUH in 2014–2015 were included in this study. Demographic information and medical history were obtained from patients’ electronic medical records, which are prospectively maintained. This study was reviewed and approved by the ethical research committee of the Faculty of Medicine at King Abdulaziz University under the reference number (235–15). In total, 155 nonduplicate clinical isolates of E. faecalis were analyzed in this study.
Identification and antimicrobial susceptibility screening
The purified isolates were freshly cultured on Columbia blood agar plates at 37 °C for 20 h using a biosafety level-2 cabinet and stored at − 80 °C in 15% glycerol and 1% skim milk. Isolates were identified by high-throughput MALDI-TOF using a VITEK-MS (bioMérieux, France) system following the manufacturer’s protocol [15]. The calibration was performed using standard Escherichia coli ATCC 25922 to validate the run. All isolates were tested for antimicrobial susceptibility using an automated VITEK-2 (bioMérieux) system with a specific AST-GP2 card. VITEK 2 system used broth microdilution minimum inhibitory concentration method for susceptibility testing and perform repetitive turbidimetric monitoring of bacterial growth during an abbreviated incubation period. MIC results were interpreted based on the Clinical and Laboratory Standards Institute guidelines [16]. The criteria of Magiorakos et al. was used to defined MDR isolates [17].
Amplification of the AMR genes
Genes responsible for AMR to vancomycin were amplified from the phenotypically resistant E. faecalis isolates using primers, and PCR conditions described previously [18]. Gel-purified amplified PCR products were sequenced with ABI prism sequencer 3730 (Applied Biosystems, USA). NCBI nucleotide BLAST was used to confirm the amplification of the respective genes.
PFGE and whole genome sequencing
Genotyping of the E. faecalis isolates was performed using PFGE in a CHEF-DR II apparatus (Bio-Rad, USA) as previously described [19], with some modifications. The optical density of an E. faecalis suspension was adjusted to 1 at 600 nm. The SmaI (60 units) restriction enzyme (Thermo Fisher Scientific, USA) was used for digestion of the plugs. PFGE was performed for 24 h at 6 V, with an initial switch of 3.5 s and the final switch at 23.5 s. SmaI-digested S. aureus strain NCTC8325 was used as a DNA molecular size control. Forty-four representative isolates from PFGE bands patterns were selected for whole genome sequencing. Genomic DNA was extracted from E. faecalis isolates using UltraClean® Microbial DNA isolation kit (MO BIO Laboratories, Inc. USA). Genomic libraries were prepared using Nextera XT DNA library preparation kit (Illumina, Inc., USA) and sequencing was performed using V3, 2 × 300 bp chemistry on a MiSeq platform (Illumina, Inc., USA).
Data analysis
BioNumerics software (V 7.6.0) from Applied Maths was used to analyze the bands patterns from PFGE, and a dendrogram was constructed using unweighted pair group with arithmetic mean (UPGMA). The default parameters of a 1% tolerance and an 85% similarity index were used for clustering the isolates.
The generated reads from whole genome sequencing were filtered according to the read qualities. The genome assemblies were prepared with SPAdes 3.9 algorithm, and sequence reads were mapped to clinical isolate E. faecalis reference genome V583. Single nucleotide polymorphisms (SNPs) were determined in the core genomes and were used to construct a maximum likelihood phylogenetic tree using CSI Phylogeny 1.4 [20]. The phylogenetic tree was visualized using Interactive Tree of Life (iTOL) tool [21]. ARGs were identified using ResFinder3.1 [22], ARG-ANNOT (Antibiotic Resistance Gene-ANNOTation) [23], and CARD (Comprehensive Antibiotic Resistance Database) [24]. VirulenceFinder 2.0 was used to identify virulence-associated genes [25]. Insertion sequences were identified using ISfinder [26, 27] and were reconfirmed from the BLASTn at NCBI. To track the epidemiology in a global context, the sequence types (STs) of the isolates were identified using an MLST scheme based on seven housekeeping genes [9]. The similarities between different STs were investigated using BioNumerics (V 7.6.0) with UPGMA and categorical coefficient of similarity. STs were grouped into clonal complexes based on single and double locus variants using eBburst (http://eburst.mlst.net). Pearson chi-square or likelihood ratio was used with a threshold of p < 0.05 for statistical significance. SPSS version 22 was used for statistical analysis. Genome sequences were deposited into the European Nucleotide Archive (ENA) under accession number ERS2489758-ERS2489715.
Results
Demographic and clinical characteristics
The majority of the isolates were recovered from inpatients (n = 102, 65.8%). In 75.5% of cases, infection was community acquired, and in 24.5% of cases, infection was probably hospital acquired. In the latter cases, infection was detected in patients after 72 h of admission in the hospital (Additional file 1: Table S1). Isolates were recovered from patients with various diseases, which were broadly classified as malignancy, renal and kidney-associated disease, urinary tract infection (UTI), gastrointestinal tract infection, respiratory tract infection, and pregnancy (Additional file 1: Figure S1). Healthcare-associated infection was detected in eight patients with a malignancy. Community-acquired infection with E. faecalis was most frequently observed in patients with chronic kidney disease and renal failure (n = 22, 14.2%) or UTI (n = 13, 8.4%) and in pregnant patients (n = 13, 8.4%) (Additional file 1: Figure S1). The majority of the isolates were recovered from patients who were > 50 years old (n = 67, 43.2%). Importantly, 23 (14.8%) of the E. faecalis isolates were recovered from neonates (≤1 year), and 19 of whom were inpatients (Additional file 1: Table S1). The isolates were recovered from heterogeneous clinical specimens, mostly urine-midstream (n = 58, 37.4%) and urine-catheter (n = 49, 31.6%) followed by wound swab (n = 13, 8.4%) and blood (n = 11, 7.1%). The strains were mainly isolated from Saudi patients (n = 81, 52.3%) and expatriates representing 16 different nationalities (Additional file 1: Figure S2). Other demographic information is presented in Additional file 1: Table S1.
Antimicrobial susceptibility analysis
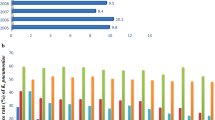
The majority of the E. faecalis isolates (96.1%) had an MDR pattern. In total, 60% of the isolates were resistant to ≥5 tested antibiotics, and eight isolates were resistant to ≥10 antibiotics from different classes and mainly were from macrolide, quinolone, tetracycline, lincosamide, and streptogramin. The isolates Efs236 and Efs249, recovered from wound swab and urine-midstream, were found to be resistant to 11 and 12 antibiotics, respectively. Ampicillin resistance was detected in 10 isolates with a MIC range of 8–32 μg/ml. Six of the isolates were resistant to vancomycin (MIC ≥32 μg/ml). The highest resistance was observed against clindamycin (99.3%). More than 85% of the isolates were resistant or intermediately resistant to erythromycin, quinupristin/dalfopristin, and tetracycline (Fig. 1). Forty-nine percent of the isolates were resistant to high-dose gentamycin, and 38.7% were resistant to high-dose streptomycin (Fig. 1). For the quinolone group, 43.2% isolates were resistant to moxifloxacin and levofloxacin, whereas 34.2% isolates were resistant or intermediately resistant to ciprofloxacin. Four of the isolates were resistant to linezolid. All the isolates were susceptible to tigecycline (Fig. 1).

Antimicrobial resistance and susceptibility profile of the tested antibiotics against E. faecalis isolates. The x-axis values are expressed in percentage
Evaluation of AMR pattern
In total, 37 different patterns of antibiotic resistance were recorded for 155 isolates, including 13 groups and 24 unique patterns (Additional file 1: Figure S3). The most dominant pattern (P1) of resistance involved four antibiotics (tetracycline, erythromycin, clindamycin, and quinupristin/dalfopristin) and was observed in 38 isolates, followed by the P2 pattern of resistance to nine antibiotics in 26 isolates. Antibiotype pattern P3 was identified in 18 isolates that harbored resistance to five antibiotics. Antibiotype pattern P13 was detected in two isolates that were resistant to 12 antibiotics (Additional file 1: Figure S3).
PCR analysis of vancomycin resistance genes
Phenotypically vancomycin resistant isolates were further characterized for the respective van genes. Vancomycin-resistant isolates mainly carried the vanA gene (5/6), and one isolate was positive for vanB; vanC1/C2 was not detected in any of the vancomycin-resistant isolates. The Efs251 isolate showed PCR amplification of the vanA gene cluster.
PFGE and genome analysis
PFGE revealed substantial heterogeneity in the E. faecalis isolates. The isolates were clustered into 44 clades at 85% similarity index in the UPGMA dendrogram constructed from the bands patterns (Additional file 1: Figure S4). A representative isolate from each clade underwent genome sequencing. Genomes of the 44 isolates covered 70.6% of the genome of the reference strain V583. The individual isolates covered the reference genome within a range of 2,501,623 to 2,987,577 nucleotides. A phylogenetic tree based on SNPs grouped 44 isolates into three clusters and two minor clusters (Fig. 2). Annotation of the tree with geographical region indicated clusters 1, 3, 4, and 5 were mainly found in the patients from the Middle East, whereas cluster 2 isolates were found in patients of different nationalities (Fig. 2).

Phylogenetic linkage among E. faecalis isolates based on SNPs in core genomes. Colored branches indicate the three dominant lineages and the outer colored ring indicates the nationality
MLST analysis and identification of clonal complex
We assigned sequence types (STs) to the genome sequences of the 44 isolates, which accounted for 17 STs, including two novels STs (ST862 and ST863) based on new combinations of the known alleles (Fig. 3). In addition, all 17 STs were reported for the first time from Saudi Arabia and showed global distribution (Fig. 3). Eleven isolates were grouped together under a single dominant sequence type ST179 (25%, n = 11), followed by ST16 (18.2%, n = 8). Other STs were identified in ≤5 isolates. Isolates from clusters 1 and 2 from SNPs tree were assigned to ST179 and ST16, respectively (Fig. 2). Clusters 3, 4, and 5 were assigned to ST28, ST480, and ST128, respectively. eBURST resolved 17 isolates with a unique allelic profile into one group and 15 singletons at single-locus and double-locus variants (Additional file 1: Figure S5). The blast with MLST database through eBURST grouped the identified STs into 14 distinct clonal complexes (CC) including major CC16, which comprised ST16 and single-locus variant ST179 followed by ST6 from CC2 (Additional file 1: Figure S5). In total, 12 different STs were detected in Saudi patients, including a novel ST862 recovered from pleural fluid (Fig. 4). The second novel ST863 was identified from urine-midstream of an Egyptian patient. Five STs were common between Saudi and expatriate populations (Fig. 4). ST16 and ST480 were recovered mainly from urine-midstream and urine-catheter samples, whereas other STs were recovered from heterogeneous clinical samples (Fig. 4). No significant association was observed between STs and diagnosed disease type; for example, ST6, ST16, and ST480 were detected in samples from multiple hospital units and patients with different diseases. However, the ST179 isolates were found at relatively higher numbers in the pediatric ward. All isolates of ST16 and ST179 from CC16 and ST6 from CC2 were resistant to high-dose gentamicin (Fig. 4). However, the ST179 isolates were susceptible to the tested antibiotics from the quinolone group (Fig. 4). A similar pattern of resistance against nine antibiotics was observed in the ST480 isolates. No specific prevalence of vancomycin-resistant isolates was noticed in any ST.

Global distribution of E. faecalis STs identified in Saudi Arabia. New STs are presented in red color, whereas other STs were found in different countries

UPGMA dendrogram from the pattern of pairwise differences in alleles that revealed the genetic relationships of STs among the E. faecalis isolates, along with the country (Ctry), infection (Inf), specimen source, and antibiotic resistance (AR) information. HA, hospital-acquired; CA, community-acquired; UC, urine-catheter; UM, urine-midstream; Bl, blood; WS, wound swab; TAS, tracheal aspirate; TS, tissue swab; PF, peritoneal fluid
ARGs and insertion sequence families
In total, 34 ARGs and variants were detected in the genome-sequenced isolates. The frequency of ARGs was found to be in the range of 5–24 genes, whereas nine isolates predominantly carried 13 ARGs followed by six isolates that harbored seven ARGs (Fig. 5). A maximum of 24 ARGs were found in the isolate EFs93 and 22 ARGs in EFs208. Two other isolates had 20 ARGs (Fig. 5). Moreover, aminoglycoside, trimethoprim, and efflux pump-mediated resistance genes were found in most of the isolates (Additional file 1: Figure S6). Aminoglycoside resistance was conferred by the presence of nine resistance genes, while aph(3′)-III gene was found in 27 isolates and aac(6′)-aph(2″) and aad(6) were found in ≥25 isolates (Additional file 1: Figure S6). Streptomycin resistance was conferred by the most common genes, ant(6)-Ia, which was detected in 29 isolates. Macrolide and streptogramin-resistance gene isaA was found in all sequenced isolates, while mphD and erm(B) genes were found in 42 and 30 isolates, respectively (Additional file 1: Figure S6). Most of the isolates were carrying efflux-associated ARGs (efrA, emeA, and efrB). Among the three trimethoprim-resistance genes, dfrE and dfrG were detected in 42 and 9 genomes, respectively. Tetracycline-resistance tet(M) gene was found in 38 genomes (Additional file 1: Figure S6). One isolate demonstrated horizontally acquired resistance gene optrA, which is responsible for linezolid resistance.

Antimicrobial resistance genes patterns identified from genome sequencing of 44 E. faecalis isolates. Total 34 resistance genes and variants were found in the analyzed genomes
Three families of insertion sequences (ISs) IS30 (IS6770, IS1062, and ISEnfa364), IS256 (ISEf1 and IS16), and IS1182 (ISEnfa2) were identified in the E. faecalis genomes. Distribution of the IS families and their members are summarized in Additional file 1: Table S2, illustrating their abundance and association with E. faecalis STs. Family IS30 (n = 24) was found at a relatively higher frequency, followed by IS256 (n = 16) and IS1182 (n = 3).
Virulence-associated genes analysis
A total of 22 putative virulence genes were identified, among which sex pheromone (cOB1, cad, and cCF10), Sortase (srtA), endocarditis and biofilm-associated pili genes (ebpB, ebpA, and ebpC), adhesin (efaAfs, and ace), thiol peroxidase (tpx), and E. faecalis virulence factor ElrA genes were commonly detected in ≥97.7% genomes from 44 isolates (Additional file 1: Figure S7). Hyalurodinase gene hylA was identified in 38 genomes. Overall, 14–22 virulence-associated genes were detected in the analyzed genomes. Cytolysin toxin-associated genes (cylA, cylB, cylL, and cylM) were more prevalent among ST6, ST16, ST28, and ST179.
Discussion
Consistent with the international trend, the prevalence of enterococcal infections is increasing in Saudi Arabia, and E. faecalis is one of the most common enterococcal species isolated from hospital-associated infections. The ECDC has estimated that enterococci are responsible for 8% of the healthcare-associated infections on average in Europe, and the agency has placed them in the category of pathogens posing a major threat to healthcare systems [4]. Enterococcal species are intrinsically resistant to a broad range of antibiotics, such as cephalosporins and sulfonamides [28]. They represent a major infection control challenge because of their ability to acquire additional resistance through the transfer of plasmids and transposons and because they can disseminate easily in the hospital environment.
In this study, linezolid, tigecycline, and vancomycin demonstrated > 95% activity against E. faecalis isolates in vitro. The antibiotics quinupristin/dalfopristin, clindamycin, and erythromycin demonstrated almost no coverage, and other antibiotics such as high-dose streptomycin, gentamycin, and ciprofloxacin demonstrated suboptimal coverage. Previous studies from Saudi Arabia revealed 21–25% resistance to high-dose gentamycin and 11–13% resistance to high-dose streptomycin [29, 30]. The high level of aminoglycoside resistance observed in this study is highly concerning, given that aminoglycosides are used in combination with other active molecules, mainly β-lactams, to treat enterococcal infections such as enterococcal endocarditis [28]. In particular, gentamycin is used as a synergistic antibiotic with ampicillin [31], and amoxicillin is the first choice of treatment for E. faecalis causing UTIs [32]. In contrast to a study from the eastern region of Saudi Arabia, comparatively lower resistance to ampicillin and higher resistance to erythromycin, gentamycin, streptomycin, and tetracycline were observed in this study, suggesting the diverse geographical distribution of MDR E. faecalis isolates in Saudi Arabia [30, 33].
The rate of vancomycin-resistant E. faecalis in this study was slightly higher than that found in a previous study from Saudi Arabia that was conducted at King Khalid Hospital in Riyadh. That study identified the vancomycin-resistant phenotype in 0–1.8% of isolates [30]. The resistance rates reported by the National Healthcare Safety Network during 2009–2010 was between 6.2% and 9.8% for E. faecalis, depending on the site of infection [34]. Despite the increasing number of reports of VRE in different geographical regions of the world, there is a distinct lack of data regarding the molecular characterization of VRE isolates originating from the Middle East region. No genotypic characterization of vancomycin-resistance genes and other acquired AMR genes in E. faecalis has been described in the available literature from Saudi Arabia. In this study, acquired vanA and vanB genes were identified in the vancomycin-resistant E. faecalis isolates from patients in the western region of Saudi Arabia. No intrinsic resistance genes (vanC1/C2) were detected in the tested isolates. Similar results have been reported in Europe, where a mix of VRE carrying vanA and vanB were found [6]. Enterococcus faecalis isolates tested in this study also possessed gyrA, and parC, genes conferring resistance to quinolone groups of antibiotics including ciprofloxacin, levofloxacin, and moxifloxacin that are commonly prescribed for UTIs, enteric infections, and respiratory tract infections.
Consistent with previous studies, the most common aminoglycoside-modifying enzyme genes (aac(6′)-Ie-aph(2″)-Ia and ant(6′)-Ia) were found in the E. faecalis isolates that were resistant to high-dose gentamycin and streptomycin [35]. Two mechanisms are responsible for the cross resistance to macrolide-lincosamide-streptogramin A in E. faecalis, including an intrinsic lsa gene and a change in the target site of erythromycin that is mediated by the erm(B) gene [36, 37]. In this study, we found the lsa gene in all genome sequence isolates, and the erm(B) gene was found in 68.1% isolates, similar to previous studies from the United States, China, and Korea [36, 38, 39]. The results obtained in this study are consistent with a previous report indicating that 98% of the E. faecalis isolates possess emeA gene and ATP-binding cassettes (ABC), which are efrA and efrB [40].
The ability of enterococci to form biofilm contributes to the pathogenicity of the bacteria in nosocomial infections because mature biofilms of E. faecalis can withstand antimicrobial agents up to 100- to 1000-fold concentrations. In contrast to previous studies, a large group of virulence factors was found in the genome sequences of E. faecalis isolates, as seen in other Gram-positive cocci, such as β-hemolytic Streptococcus and S. aureus. In this study, 22 virulence genes were retrieved from the E. faecalis isolates, including the genes associated with biofilm formation as previously mentioned. The presence of virulence determinants in enterococcal isolates assist them in acquiring adaptive elements that provide them with evolutionary benefits for relative fitness in hospital settings [41, 42]. In comparative studies, E. faecalis is considered to be inherently more virulent and to have greater capability to acquire virulence factors than E. faecium strains [1]. Overall, variation was observed in the relative distribution of virulence factors in the enterococcal species from different geographical regions [43, 44]. Findings from the current study are consistent with a previous report that esp and cylA genes were mainly found in CC16 isolates. Importantly, the cCF10 gene, which activates the conjugation of pCF10 plasmid, was found in this study. This plasmid plays an important role in the dissemination of virulence factors and resistance genes among enterococci [45].
A noticeable diversity of strains was observed in the E. faecalis isolates from Saudi Arabia, and 17 distinct STs, including two novel STs, from the genome sequences of 44 isolates were identified. Major clusters from a phylogenetic tree based on SNPs were assigned to ST179 and ST16 from CC16. Generally, STs from CC16 are considered to be well acclimated to hospital environments. They have previously been reported to cause human infections, acquire exogenous genes via recombination, and be able to carry vanA or vanB genes, conjugative plasmids, and transposons involved in the genetic transfer of resistance and virulence in hospital-derived isolates [46,47,48]. All 18 isolates belonging to CC16 from various specimen types in this study presented MDR phenotypes and genotypes. In addition, they carried virulence genes, most commonly gelE and asa1, which accords with a previous study [49]. In contrast to previous reports, CC2 isolates were vancomycin sensitive, which supports the hypothesis that this clone was originally vancomycin susceptible and later subsets acquired the vancomycin-resistance gene. In addition, several STs such as ST21, ST480, and ST40 detected in this study were previously reported in China, Tunisia, France, and Spain from human subjects, hospitalized patients, and wastewater, and shared the same characteristics in terms of high-dose antibiotic-resistant phenotypes, resistance genotypes, and virulence genes [50,51,52]. The lack of information on the population structure of E. faecalis in neighboring countries makes it difficult to speculate about the regional spread of these novel STs and other STs detected in Saudi Arabia.
Conclusion
This study provided the first insights into the population structure of E. faecalis isolates from healthcare facilities in the western region of Saudi Arabia. The threat of antimicrobial-resistant E. faecalis is steadily increasing, with diverse clonal composition and domination by lineages associated with nosocomial infection in Saudi Arabia. MDR isolates of E. faecalis acquired an increased number of virulence gene and diverse patterns of ARGs. Evaluation of antimicrobial susceptibility suggests that ampicillin, tigecycline, and linezolid could be used as treatment options for combatting aminoglycoside- and macrolide-resistant E. faecalis strains in healthcare facilities in Saudi Arabia. The appearance of new STs in the studied hospital could be a warning about the emergence and rapid evolution of this clinically important resistant bacteria, and it suggests the necessity of active surveillance in other hospitals and areas of the country.
Abbreviations
- AMR:
-
Antimicrobial resistance
- ARG-ANNOT:
-
Antibiotic resistance Gene-ANNOTation
- ARGEs:
-
Antimicrobial resistance genes
- CARD:
-
Comprehensive antibiotic resistance database
- CC:
-
Colonel complex
- ECDC:
-
European centre for disease prevention and control
- ENA:
-
European Nucleotide Archive
- KAUH:
-
King Abdulaziz university hospital
- MALDI-TOF:
-
Matrix assisted laser desorption ionization - time of flight
- MDR:
-
Multidrug-resistant
- MIC:
-
Minimum inhibitory concentration
- MLST:
-
Multilocus sequence typing
- PFGE:
-
Pulsed-field gel electrophoresis
- SNPs:
-
Single nucleotide polymorphisms
- ST:
-
Seventeen sequence type
- UPGMA:
-
Unweighted pair group with arithmetic mean
- UTI:
-
Urinary tract infection
- VRE:
-
Vancomycin-resistant enterococci
References
Upadhyaya PG, Ravikumar K, Umapathy B. Review of virulence factors of enterococcus: an emerging nosocomial pathogen. Indian J Med Microbiol. 2009;27(4):301.
Gordon S, Swenson JM, Hill B, Pigott N, Facklam R, Cooksey R, Thornsberry C, Jarvis W, Tenover F. Antimicrobial susceptibility patterns of common and unusual species of enterococci causing infections in the United States. Enterococcal study group. J Clin Microbiol. 1992;30(9):2373–8.
Moellering RC Jr. Emergence of Enterococcus as a significant pathogen. Clin Infect Dis. 1992:1173–6.
European Centre for Disease Prevention and Control. Point prevalence survey of health care associated infections and antimicrobial use in European acute care hospitals. Stockholm: ECDC; 2013. https://ecdc.europa.eu/sites/portal/files/media/en/publications/Publications/healthcare-associated-infections-antimicrobial-use-PPS.pdf.
Arias CA, Contreras GA, Murray BE. Management of multidrug-resistant enterococcal infections. Clin Microbiol Infect. 2010;16(6):555–62.
Werner G, Coque T, Hammerum A, Hope R, Hryniewicz W, Johnson A, Klare I, Kristinsson K, Leclercq R, Lester C: Emergence and spread of vancomycin resistance among enterococci in Europe. 2008.
Ruoff KL, De La Maza L, Murtagh MJ, Spargo JD, Ferraro MJ. Species identities of enterococci isolated from clinical specimens. J Clin Microbiol. 1990;28(3):435–7.
Homan WL, Tribe D, Poznanski S, Li M, Hogg G, Spalburg E, van Embden JD, Willems RJ. Multilocus sequence typing scheme for Enterococcus faecium. J Clin Microbiol. 2002;40(6):1963–71.
Ruiz-Garbajosa P, Bonten MJ, Robinson DA, Top J, Nallapareddy SR, Torres C, Coque TM, Cantón R, Baquero F, Murray BE. Multilocus sequence typing scheme for Enterococcus faecalis reveals hospital-adapted genetic complexes in a background of high rates of recombination. J Clin Microbiol. 2006;44(6):2220–8.
Rice HE, Brown RL, Gollin G, Caty MG, Gilbert J, Skinner MA, Glick PL, Azizkhan RG. Results of a pilot trial comparing prolonged intravenous antibiotics with sequential intravenous/oral antibiotics for children with perforated appendicitis. Arch Surg. 2001;136(12):1391–5.
Courvalin P. Vancomycin resistance in gram-positive cocci. Clin Infect Dis. 2006;42(Supplement_1):S25–34.
Woodford N. Epidemiology of the genetic elements responsible for acquired glycopeptide resistance in enterococci. Microb Drug Resist. 2001;7(3):229–36.
Chang S, Sievert DM, Hageman JC, Boulton ML, Tenover FC, Downes FP, Shah S, Rudrik JT, Pupp GR, Brown WJ. Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N Engl J Med. 2003;348(14):1342–7.
Salem-Bekhit MM, Moussa IM, Muharram MM, Alanazy FK, Hefni HM. Prevalence and antimicrobial resistance pattern of multidrug-resistant enterococci isolated from clinical specimens. Indian J Med Microbiol. 2012;30(1):44–51.
Wattal C, Oberoi J, Goel N, Raveendran R, Khanna S. Matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS) for rapid identification of micro-organisms in the routine clinical microbiology laboratory. Eur J Clin Microbiol Infect Dis. 2017;36(5):807–12.
CLSI. Performance standards for antimicrobial susceptibility testing; 26 ed. Wayne, PA: Clinical and Laboratory Standards Institute; 2016.
Magiorakos AP, Srinivasan A, Carey R, Carmeli Y, Falagas M, Giske C, Harbarth S, Hindler J, Kahlmeter G, Olsson-Liljequist B. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2012;18(3):268–81.
Miele A, Bandera M, Goldstein BP. Use of primers selective for vancomycin resistance genes to determine van genotype in enterococci and to study gene organization in VanA isolates. Antimicrob Agents Chemother. 1995;39(8):1772–8.
Nallapareddy SR, Duh R-W, Singh KV, Murray BE. Molecular typing of selected Enterococcus faecalis isolates: pilot study using multilocus sequence typing and pulsed-field gel electrophoresis. J Clin Microbiol. 2002;40(3):868–76.
Kaas RS, Leekitcharoenphon P, Aarestrup FM, Lund O. Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS One. 2014;9(8):e104984.
Letunic I, Bork P. Interactive tree of life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2006;23(1):127–8.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC bioinformatics. 2009;10(1):421.
Gupta SK, Padmanabhan BR, Diene SM, Lopez-Rojas R, Kempf M, Landraud L, Rolain J-M. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother. 2014;58(1):212–20.
Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, Lago BA, Dave BM, Pereira S, Sharma AN. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2016:gkw1004.
Joensen KG, Scheutz F, Lund O, Hasman H, Kaas RS, Nielsen EM, Aarestrup FM. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J Clin Microbiol. 2014;52(5):1501–10.
Carattoli A, Zankari E, García-Fernández A, Larsen MV, Lund O, Villa L, Aarestrup FM, Hasman H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 2014;58(7):3895–903.
Siguier P, Pérochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34(suppl_1):D32–6.
Kristich CJ, Rice LB, Arias CA: Enterococcal infection—treatment and antibiotic resistance. 2014.
El-Kersh TA, Marie MA, Al-Sheikh YA, Al-Agamy MH, Al-Bloushy AA. Prevalence and risk factors of early fecal carriage of Enterococcus faecalis and Staphylococcus spp and their antimicrobial resistant patterns among healthy neonates born in a hospital setting in Central Saudi Arabia. Saudi Med J. 2016;37(3):280.
Salem-Bekhit MM, Moussa IMI, Elsherbini MMAM, AlRejaie S. Increasing prevalence of high-level gentamicin resistant enterococci: an emerging clinical problem. Afr J Microbiol Res. 2011;5(31):5713–20.
Fernández-Hidalgo N, Almirante B, Gavaldà J, Gurgui M, Peña C, de Alarcón A, Ruiz J, Vilacosta I, Montejo M, Vallejo N. Ampicillin plus ceftriaxone is as effective as ampicillin plus gentamicin for treating Enterococcus faecalis infective endocarditis. Clin Infect Dis. 2013;56(9):1261–8.
Richey E, Waters P, Jovic M, Rakhman C. Treatment of ampicillin-resistant enterococcus faecium urinary tract infections. Fed Pract. 2015;32:20–3.
Zeyaullah M, Kaul V. Prevalence of urinary tract infection and antibiotic resistance pattern in Saudi Arabia population. Glob J Biol, Agric Health Sci. 2015;4(1):206–14.
Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, Kallen A, Limbago B, Fridkin S. Antimicrobial-resistant pathogens associated with healthcare-associated infections summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect Control Hosp Epidemiol. 2013;34(1):1–14.
Padmasini E, Padmaraj R, Ramesh SS. High level aminoglycoside resistance and distribution of aminoglycoside resistant genes among clinical isolates of Enterococcus species in Chennai, India. Sci World J. 2014;2014.
Singh KV, Weinstock GM, Murray BE. An Enterococcus faecalis ABC homologue (Lsa) is required for the resistance of this species to clindamycin and quinupristin-dalfopristin. Antimicrob Agents Chemother. 2002;46(6):1845–50.
Portillo A, Ruiz-Larrea F, Zarazaga M, Alonso A, Martinez JL, Torres C. Macrolide Resistance Genes inEnterococcus spp. Antimicrob Agents Chemother. 2000;44(4):967–71.
Jia W, Li G, Wang W. Prevalence and antimicrobial resistance of Enterococcus species: a hospital-based study in China. Int J Environ Res Public Health. 2014;11(3):3424–42.
Lee E-W, Huda MN, Kuroda T, Mizushima T, Tsuchiya T. EfrAB, an ABC multidrug efflux pump in Enterococcus faecalis. Antimicrob Agents Chemother. 2003;47(12):3733–8.
Sanchez Valenzuela A, Lavilla Lerma L, Benomar N, Gálvez A, Perez Pulido R, Abriouel H. Phenotypic and molecular antibiotic resistance profile of Enterococcus faecalis and Enterococcus faecium isolated from different traditional fermented foods. Foodborne Pathog Dis. 2013;10(2):143–9.
Toledo-Arana A, Valle J, Solano C, MaJ A, Cucarella C, Lamata M, Amorena B, Leiva J, Penadés JR, Lasa I. The enterococcal surface protein, Esp, is involved in Enterococcus faecalis biofilm formation. Appl Environ Microbiol. 2001;67(10):4538–45.
Guiton PS, Hung CS, Kline KA, Roth R, Kau AL, Hayes E, Heuser J, Dodson KW, Caparon MG, Hultgren SJ. Contribution of autolysin and sortase a during Enterococcus faecalis DNA-dependent biofilm development. Infect Immun. 2009;77(9):3626–38.
Garg S, Mohan B, Taneja N. Biofilm formation capability of enterococcal strains causing urinary tract infection Vis-a-Vis colonisation and correlation with enterococcal surface protein gene. Indian J Med Microbiol. 2017;35(1):48.
Seno Y, Kariyama R, Mitsuhata R, Monden K, Kumon H. Clinical implications of biofilm formation by Enterococcus faecalis in the urinary tract. Acta Med Okayama. 2005;59(3):79–87.
Dunny G. Genetic functions and cell–cell interactions in the pheromone-inducible plasmid transfer system of Enterococcus faecalis. Mol Microbiol. 1990;4(5):689–96.
Ben HY, Chairat S, Hamdi N, Gharsa H, Ben RS, Ceballos S, Torres C, Ben KS. Antimicrobial resistance and genetic lineages of faecal enterococci of wild birds: emergence of vanA and vanB2 harbouring Enterococcus faecalis. Int J Antimicrob Agents. 2018.
Freitas AR, Novais C, Ruiz-Garbajosa P, Coque TM, Peixe L. Clonal expansion within clonal complex 2 and spread of vancomycin-resistant plasmids among different genetic lineages of Enterococcus faecalis from Portugal. J Antimicrob Chemother. 2009;63(6):1104–11.
Oravcova V, Zurek L, Townsend A, Clark AB, Ellis JC, Cizek A, Literak I. A merican crows as carriers of vancomycin-resistant enterococci with vanA gene. Environ Microbiol. 2014;16(4):939–49.
Tedim AP, Ruiz-Garbajosa P, Corander J, Rodríguez CM, Cantón R, Willems RJ, Baquero F, Coque TM. Population biology of intestinal Enterococcus isolates from hospitalized and nonhospitalized individuals in different age groups. Appl Environ Microbiol. 2015;81(5):1820–31.
Quinones D, Kobayashi N, Nagashima S. Molecular epidemiologic analysis of Enterococcus faecalis isolates in Cuba by multilocus sequence typing. Microb Drug Resist. 2009;15(4):287–93.
Cai J, Wang Y, Schwarz S, Lv H, Li Y, Liao K, Yu S, Zhao K, Gu D, Wang X. Enterococcal isolates carrying the novel oxazolidinone resistance gene optrA from hospitals in Zhejiang, Guangdong, and Henan, China, 2010–2014. Clin Microbiol Infect. 2015;21(12):1095. e1091–4.
Zischka M, Künne CT, Blom J, Wobser D, Sakιnç T, Schmidt-Hohagen K, Dabrowski PW, Nitsche A, Hübner J, Hain T. Comprehensive molecular, genomic and phenotypic analysis of a major clone of Enterococcus faecalis MLST ST40. BMC Genomics. 2015;16(1):175.
Acknowledgments
The results were obtained by BioNumerics evaluation license from Applied Maths, and we have received permission to publish. The authors also acknowledge with thanks the Science and Technology Unit, King Abdulaziz University for their technical support.
Funding
This project was funded by the National Plan for Science, Technology and Innovation (MAARIFAH) - King Abdulaziz City for Science and Technology - the Kingdom of Saudi Arabia - award number (12MED3108–03).
Availability of data and materials
Genome sequences were deposited into the European Nucleotide Archive (ENA) under accession number ERS2489758-ERS2489715.
Author information
Authors and Affiliations
Contributions
MF and MY: contributed to the experimental work, data analysis and manuscript drafting. MY, RA and EA: conception and design of the study. SA: contributed to the experimental work. AF and MA: contributed in samples collection and acquisition of the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethical research committee of the Faculty of Medicine at King Abdulaziz University (HA-02-J-008), under the reference number (235–15) and performed according to the approved guidelines. Formal consent was therefore not required.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. Percentage distribution of hospital-acquired infection (HA), and community-acquired infection (CA) infections of E. faecalis in different disease patients. Figure S2. Distribution of the E. faecalis isolates analyzed in this study based on nationalities. The number of isolates is mentioned with the country name. Figure S3. Heat map of antimicrobial resistance and susceptibility patterns among the 155 E. faecalis isolates. Figure S4. UPGMA dendrogram of pulsed-field gel electrophoresis (PFGE) bands patterns from the clinical isolates of E. faecalis. Figure S5. eBURST analysis of the STs from E. faecalis isolates. The pink nodes indicate the STs detected in this study that were present in the MLST database. Green nodes indicate the novel STs detected in this study and red circled. Figure S6. Distribution of antimicrobial resistance genes among clinical E. faecalis isolates. Total 34 ARGs and variants were retrieved from the genomes sequence of the 44 isolates. The y-axis values are expressed in number. Figure S7. Distribution of virulence genes retrieved from the genomes sequence of E. faecalis isolates. Total 22 virulence-associated genes were identified in this study, and among them, six genes (cOB1, SrtA, ebpB, ebpA, efaAfs, and ace) were commonly found in the 44 isolates. The y-axis values are expressed in number. Table S1. Distribution of E. faecalis isolates according to demographic and clinical data. Table S2. Distribution of insertion sequences (ISs) in the E. faecalis genomes. (PDF 882 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Farman, M., Yasir, M., Al-Hindi, R.R. et al. Genomic analysis of multidrug-resistant clinical Enterococcus faecalis isolates for antimicrobial resistance genes and virulence factors from the western region of Saudi Arabia. Antimicrob Resist Infect Control 8, 55 (2019). https://doi.org/10.1186/s13756-019-0508-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13756-019-0508-4