
Abstract
T helper type 2 (TH2) cells were previously thought to be the main initiating effector cell type in asthma; however, exaggerated TH2 cell activities alone were insufficient to explain all aspects of asthma. Asthma is a heterogeneous syndrome comprising different phenotypes that are characterized by their different clinical features, treatment responses, and inflammation patterns. The most-studied subgroups of asthma include TH2-associated early-onset allergic asthma, late-onset persistent eosinophilic asthma, virus-induced asthma, obesity-related asthma, and neutrophilic asthma. The recent discovery of human innate lymphoid cells capable of rapidly producing large amounts of cytokines upon activation and the mouse data pointing to an essential role for these cells in asthma models have emphasized the important role of the innate immune system in asthma and have provided a new means of better understanding asthma mechanisms and differentiating its phenotypes.
Similar content being viewed by others

Introduction
The immune system is classically divided into two categories, innate and adaptive immunity, according to the speed and the duration of the response, and they collaborate with each other to target different agents and perform effector functions. Through recent advances in understanding the different subsets of immune system effector cells, Annunziato et al. have recently suggested a new classification [1]. They proposed that the innate and adaptive immune systems could also be generally classified into three major kinds of cell-mediated effector immunity: categorized as type 1, comprising T-bet+ IFN-γ–producing helper cells, type 2, composed of GATA-3+ lymphocytes producing interleukin-4 (IL-4), IL-5, and IL-13, and type 3, characterized by RORγt+ lymphocytes that produce IL-17 alone or in combination with IL-22 as signature cytokines [1].
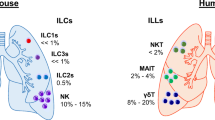
Innate immunity is known to respond quickly and without antigen specificity to signals derived from the environment or from other immune cells. Innate lymphoid cells (ILCs) are the newest described elements of the innate immune system and have received much attention over the last few years [2]. Early in the immune response, ILCs possess a lymphoid morphology, similar to adaptive B and T cells, and produce many different T helper (TH) cell cytokines but lack the recombination-activating gene (RAG)-mediated antigen specific receptors; therefore, these cells are not antigen-specific. Because ILCs are very similar to the other effector cell phenotypes, it was proposed that ILCs could be classified in a similar manner to that of TH cells. Type 1 immunity includes the IFN-γ-producing group 1 ILCs (ILC1s) that cope with intracellular pathogens through activation of mononuclear phagocytes. Group 2 ILCs (ILC2s), which secrete IL-4, IL-5, IL-9, and IL-13, are an example of Type 2 immunity. This type of immunity induces mast cell, basophil, and eosinophil activation leading to an increase in serum IgE levels and, therefore, fosters the eradication of helminthes and venoms. Group 3 ILCs (ILC3s), which are an example of type 3 immunity, produce IL-17 and/or IL-22, activate mononuclear phagocytes, recruit neutrophils, and induce epithelial antimicrobial responses, all of which help protect against extracellular fungal and bacterial infections [1]. This group includes lymphoid tissue inducer (LTi) cells that promote the formation of lymph nodes [3].
In general, ILCs constitute a distinct element of the innate immune system, providing an initial host response via specific cytokines after sensing external stimuli on the frontline. The initial priming of immune responses to pathogenic challenges is executed by ILCs with the capacity to rapidly secrete effector cytokines. All ILCs are developmentally related, and they all require the expression of the transcriptional repressor inhibitor of DNA binding 2 (Id2) and the common IL-2 cytokine receptor (γc) chain. Moreover, they all possess the IL-7 receptor α-chain (CD-127) [4].
The ILC lineage incorporates the classic cytotoxic natural killer (NK) cells and the non-cytotoxic ILC family [5]. Natural killer cells are also capable of responding to invading pathogens and exterior threats without the need for prior sensitization, and they function in the absence of RAG-recombined antigen receptor recognition. Beside their ability to release a variety of cytokines, they also have the capacity to kill other cells. NK cells were initially categorized into ILC1s, but recently it has been shown that these cells are different from non-cytotoxic ILCs because they undergo different developmental pathways [6, 7].
Non-cytotoxic ILCs have the capacity to rapidly respond to the environment by producing various cytokines, and their goal is to maintain homeostasis with tissue repair and remodeling. They are involved in lymphoid organ development and in resistance to pathogenic and nonpathogenic microorganisms. Non-cytotoxic ILCs also interact with mast cells, natural killer T (NKT) cells, eosinophils, epithelial cells, and macrophages, and they may configure the optimal milieu for setting up an adaptive response [8, 5].
Asthma includes complex innate and adaptive immune responses to environmental factors. For decades, researchers investigating the immune responses in asthma have focused on adaptive immunity, mostly on memory responses to antigens. Therefore, asthma was previously considered to be the airway manifestation of a TH2-driven response from adaptive immunity toward some specific triggers [9]. Today, advances in molecular technology and recent immunology studies have allowed us to understand much more about the impact of the innate immune system on the development of asthma and on its evolution. Negative results from the initial monoclonal treatment drug studies and cluster analysis have demonstrated that “asthma syndrome” covers distinct subgroups of a reversible obstructive lung disease with different clinical properties termed different “phenotypes” [10–12]. Although there is no consensus on a single phenotype classification for asthma, the most-studied subgroups include: TH2-associated with early-onset allergic asthma, late-onset persistent eosinophilic asthma, virus-induced asthma, obesity-related asthma, and neutrophilic asthma. All of these subgroups can be distinguished from each other by clinical factors, such as the patient age at disease onset and the involvement of particular biological pathways.
Understanding new innate pathways will allow for more accurate asthma phenotyping and, subsequently, will help direct us to personalized care for our asthmatic patients. In this review, we provide an updated view on the emerging roles of non-cytotoxic ILCs in different asthma phenotypes.
Review
ILC1s and its possible role in asthma phenotypes
ILC1s, formerly known as conventional NK cells, are present in mucosal tissues, express the IL-7 receptor, and rapidly secrete IFN-γ upon stimulation with IL-12 and IL-18, which are produced by macrophages and other cells. ILC1s are involved in the antiviral response and have been shown to expand in the intestines of patients with Crohn’s disease [13]. Although we now know that NK cells are developmentally different from ILC1s and that ILC1s lack cytotoxicity, these two cell types share some common properties [14]. Therefore, it is postulated that, like NK cells in a mouse model [15] and in human asthmatics [16], ILC1s might also have a role in the development of eosinophilic airway inflammation, which can be seen in most asthma phenotypes and even in the microbiota–immune interactions of asthma [17]. Intraepithelial ILC1s, another subset of ILC1s, have been found in human tonsillar tissue [18]. Unlike typical ILC1s, these cells are not stimulated with IL-12 and IL-18, but rather with IL-15.
ILC2s and early onset allergic asthma
For many years, early onset allergic asthma has been considered to be an adaptive immune response that develops after the prior sensitization phase to allergens. Airway epithelial cells are the frontline cells initially exposed to inhaled substances, and they actively collaborate with other immune cells, specifically pulmonary dendritic cells (DC) followed by M2 macrophages, to mount a TH2 response through the production of epithelial cell-derived cytokines, such as IL-33, IL-25 and thymic stromal lymphopoietin (TSLP) [8].
After recent studies questioning the requirement for antigen-specific adaptive TH cells in allergic asthma, the existence of a new class of the innate type-2 lymphocyte group, the ILC2s, has been described. ILC2s were first observed in the gut, emphasizing their physiological role against helminth infection [19–21]. Later, their presence was confirmed in various other tissues, including in the human lung [22]. ILC2s are also present in human peripheral blood, and their percentage is greater in asthma patients than it is in allergic rhinitis patients or in healthy controls [23, 24].
Following contact with certain microbial products, helminth infection, physical injury, or allergens in the airway, epithelial cells secrete TSLP, IL-25, and IL-33 [25, 26, 23, 19]. Afterwards the recruitment and activation of innate type 2 cells can initiate the immune response independently of adaptive immunity [27–29]. Lung ILC2s are an important source of IL-5, a growth and differentiation factor for eosinophils, and of IL-13, which can directly cause airway hyperreactivity (AHR). Cytokine production is followed by a progressive accumulation of eosinophils and mucus secretion. IL-13 is also crucial for the differentiation of TH2 cells from naive CD4+ T cells (Fig. 1) [22, 21, 20, 30]. Mouse studies have demonstrated a role for ILC2s in OVA-, HDM-, papain protease-, and Alternaria alternata-induced airway inflammation [31, 29, 32, 22, 33, 34]. Some of these observations are from RAG-deficient animals, which are adaptive immunity-deficient mice. Although evidence supporting this in human asthma has not been found yet, we speculate that the activation of ILC2s in the absence of T cells and B cells is enough to induce asthma-like symptoms, and that ILC2s may play a role in early onset allergic asthma.

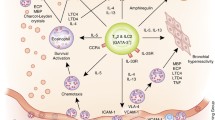
Function and regulation of group 2 lymphoid cells in different asthma phenotypes. Innate lymphoid cells group 2 (ILC2s) of early onset asthma and late onset asthma with polyposis are regulated by several elements such as the epithelial cell derived thymic stromal lymphopoietin (TSLP), interleukin 25 (IL-25) and IL-33; arachidonic acid metabolites, like prostaglandin D2 (PGD2) and leukotriene D4 (LTD4). Lung ILC2s produces IL-9 that also regulates their activation. ILC2s release IL-4, IL-5 and IL-13; then increase the airway hyperreactivity and eosinophilia. Lung ILC2s also secrete arginase 1. ILC2s can stimulate naive T cells (TH0) by IL-4, costimulatory molecules OX40L and a contact-dependent mechanism favoring TH2 polarization. In the virus induced asthma phenotype, lungs ILC2s constitute a balance between tissue repair and tissue damage via amphiregulin and type 2-cytokine secretion. The damage is potentialized by IL-33 and the repairing capacity is enhanced by maresins. Eos, eosinophil
A papain-induced asthma model showed that even in the presence of T cells, ILC2s were the major source of type 2 cytokines [22]. Another mouse model with papain-induced airway inflammation revealed that lung ILCs also produce IL-9, depending on the amount of IL-2 from the adaptive immune system, and IL-33 [35]. Moreover, a recent study showed that ILC2s in the lungs secrete arginase-1, a key enzyme in the pathophysiology of acute and chronic allergic asthma (Fig. 1) [36–38].
Being at the side that first contacts the environment, as well as the first source of type 2 cytokines, it is likely that ILC2s have a role in preparing a type 2 milieu for setting up the adaptive immune response [8]. Furthermore, major histocompatibility complex II (MHCII) is expressed on ILC2s, which provides them with the capacity for antigen presentation [39, 20]. ILC2s can promote the effector functions of CD4+ T cells via costimulatory molecules OX40L and IL-4 and by a contact-dependent mechanism favoring TH2 polarization [40, 41]. Mutually-activated ILC2s also need IL-2, possibly derived from T cells, for activation and survival [21, 20].
ILC2s and late onset asthma with nasal polyposis
Asthma onset after 12 years of age and the presence of blood eosinophilia are two important parameters for differentiating the immunologically and pathologically distinct asthma phenotype known as late onset asthma with nasal polyposis [42]. This phenotype is frequently associated with nasal polyposis and sometimes with aspirin-sensitivity [11]. Nevertheless, allergy skin test results are often positive in asthma patients with this phenotype, and even though these patients may rarely feel that their allergy symptoms were triggered by the allergens for which they tested positive [42].
Mjösberg et al. first identified ILC2s in nasal polyps of patients with rhinosinusitis (CRSwNP) [23]. Several studies have reported an increased percentage of ILC2s in the sinus mucosa of these patients compared with that in chronic rhinosinusitis patients without nasal polyps [43–45]. IL-25, IL-33, and eotaxin-3 levels, released from the sinus mucosa epithelium were also increased in CRSwNP [46]. Additionally, these patients had upregulated IL-5 and IL-13 mRNA levels [43]. The stimulation of ILC2s from human nasal polyps with TSLP has been shown to result in IL-4 release (Fig. 1) [47]. Another report found that ILC2s frequencies were associated with tissue and blood eosinophilia [45]. Additional studies focusing on the effects of ILC2s frequency on asthma control, the severity of this phenotype, and the association with the presence of aspirin sensitivity are needed.
ILC2s in virus-associated asthma and AHR
Viruses can pave the way for the development of asthma in susceptible individuals. After 2 years of age, viruses can be the trigger for a distinct phenotype of asthma known as “virus-induced asthma”. Moreover, viruses frequently provoke asthma exacerbations [48–51].
In an experimental mouse model, researchers have shown that influenza A virus can rapidly induce AHR by inducing the activation of ILCs independently of the adaptive immune system [52]. During influenza virus infection, IL-33 is released from alveolar macrophages and NKT cells, which induces ILC2 activation and the subsequent production of type 2 cytokines, IL-13 and IL-5 [52, 53]. The presence of IL-5 enables the growth and the later persistence of eosinophils, even after viral clearance. IL-5 and IL-13 are mainly responsible for the clinical symptoms of AHR. Consequently, ILC2s can promote inflammation, but they also have an opposing role during virus-induced AHR- specifically the repair of wounded lung tissue after virus infection. This effect is attained through amphiregulin, an epidermal growth factor-like growth factor (Fig. 1) [4]. The balance between the damage and repair of airways constitutes the homeostatic function of ILC2s.
Regulation of ILC2s function during asthma
Recent work on ILC2s has provided new insights into TH2-mediated asthma phenotypes, but additional questions remain. Future studies are needed to determine how this newly found source of type 2 cytokines could be regulated and how this knowledge will ameliorate our treatment options.
Role of TSLP, IL-25, and IL-33 in regulating ILC2s
Human ILC2s can be stimulated by TSLP, IL-25, and IL-33 [23, 44, 22]. Intranasal administration of IL-25 or IL-33 induces an increase in cytokine-releasing ILC2s in the lungs, bronchoalveolar lavage fluid, and mediastinal lymph nodes [31, 29, 54, 55].
-
IL-25 has an essential role in allergic airway inflammation and also in remodeling [56]. Neutralizing antibodies against IL-25 may prevent airway hyperresponsiveness in allergic asthma [57].
-
IL-33 can also activate mast cells and basophils through IgE receptors, and is a survival factor for eosinophils [58, 59]. Its effect on ILC2s is even faster and stronger than that of IL-25 [60]. These properties make IL-33 a possible target for future therapies. Like neutralizing antibodies to IL-25, neutralizing antibodies to IL-33 or to IL-33 receptor (ST2) has been shown to reduce AHR and to lessen the eosinophilic response [61].
Role of specialized pro-resolving mediators (SPM)
Asthma is an inflammatory lung disease with impaired resolution mechanisms, and understanding more about immune resolution could provide new treatments for this disease. SPM, which are essential fatty acids derived from regulating molecules, possess potent anti-inflammatory and pro-resolving capacities [62, 63]. They include lipoxins, resolvins, protectins, and maresins [64]. Investigating how ILC2s can be regulated through SPM will provide new insights into asthma pathobiology and could result in new therapeutic approaches [62].
-
Lipoxins are the leading family of SPM [63]. Lipoxin A4 might inhibit the stimulatory effects of PGD2, IL-25, and IL-33 [16].
-
Maresins are the most recently described SPM family. In a recent study, researchers demonstrated that maresins reduce lung inflammation and ILC2s expression of cytokines and increase the repairing capacity of ILC2s through amphiregulin (Fig. 1) [65]. Furthermore, regulatory T cells (Tregs) play a mandatory role in this interaction. Therefore, as potent regulators of Tregs and ILC2s, maresins may be promising therapeutic targets for asthma.
Role of leukotrienes and prostaglandins
Human ILC2s are stimulated by arachidonic acid metabolites, such as leukotrienes [32] and prostaglandins [16].
-
Lung ILC2s express receptors for cysteinyl leukotrienes, including cysteinyl leukotriene receptor 1 (CysLT1R), the high-affinity receptor for leukotriene D4 (LTD4). Following stimulation by LTD4, ILC2s produce IL-4, IL-5, and IL-13. Montelukast, a CysLT1R antagonist, can prevent the IL-5 production stimulated by leukotriene C4 and LTD4 [32].
-
Prostaglandin D2 (PGD2) is a positive regulator of ILC2s, inducing ILC2s migration and production of type 2 cytokines [16, 66]. PGD2 binds to its recently characterized receptor, Chemokine receptor, a homologous molecule expressed on T helper type 2 cells (CRTH2), which is a receptor expressed on ILC2s that is similar to a TH2 receptor [67].
Recently, a study evaluating the effect of subcutaneous grass pollen immunotherapy (SCIT) on peripheral ILC2s demonstrated that the percentage of ILC2s in untreated allergic rhinitis patients increased during pollen season, and that this percentage is correlated with the patient’s symptom scores. In contrast, the percentage of peripheral ILC2s in allergic rhinitis patients who were treated with SCIT and in control patients did not increase during pollen season [68]. An evaluation of whether this same effect occurs in allergic asthma patients remains to be conducted.
ILC3s in non-allergic asthma
Non-TH2 asthma is poorly defined and is less well understood than allergic asthma phenotypes, even though it represents a large proportion of total asthma cases [11]. This group of asthma phenotypes includes obesity-associated asthma and neutrophilic asthma.
Although the role in non-allergic asthma of type 3 immunity and IL-17, which is believed to be a TH2-released cytokine, have only recently become an area of interest, a combination of bench and bedside approaches should improve our understanding of these phenotypes [11]. Recent studies have emphasized the role of IL-17 on steroid-resistant AHR [69, 70].
ILC3s are mainly found in gut-associated lymphoid tissue (GALT) [71], but their presence in the lung has also been demonstrated [72]. They express MHC class II and are able to regulate the adaptive immune system by presenting antigens [73]. IL-23 and IL-1β rapidly stimulate ILC3s to produce IL-22, which plays a protective role through lung epithelial cells during TH2 asthma (Fig. 2) [74]. ILC3s may also produce IL-17A, which is a potent neutrophil chemotactic agent. The presence of IL-22 and IL-17A in the sputum or peripheral blood is positively correlated with the severity of asthma [75–79]. However, further studies are needed to show the role of these cytokines in non-TH2 asthma.

Mechanism of innate lymphoid cells group 3 in obesity induced asthma and their regulation. Innate lymphoid cells group 3 (ILC3s) produce interleukin 17A (IL-17A) and IL-22. Macrophages (Mϕ) produce IL-1β that engages IL-1 receptor on innate lymphoid cells group 3 (ILC3s) resulting in airway hyperreactivity. This effect can be inhibited by an IL-1 receptor (IL-1R) antagonist. ILC3s are sensitive to environmental factors, micronutrients and microbiota. Vitamin D deficiency increases ILC3s’ functions whereas Vitamin A deficiency leads to a reduction; the influence of airway microbiota on ILC3s is still unknown
Obesity-associated asthma
This asthma phenotype is difficult to control because of comorbidities and a lack of responsiveness to classic asthma treatments [11, 80]. In a mouse model of obesity-induced AHR, researchers showed a crucial role in AHR for IL-17A, which is secreted mainly from ILC3s in the absence of adaptive immunity. The same study was the first to report the presence of ILC3s in the bronchoalveolar lavage fluid of patients with lung diseases. The researchers also reported that patients with severe asthma had a higher percentage of lung IL-17-producing ILC3s, than patients with mild or no asthma. Surprisingly, a protective role for ILC2s, in which they maintain the metabolic homeostasis in obesity, has been recently demonstrated [81, 82]. This unexpected finding suggests that the role of ILC2s in obesity-associated asthma should be studied further.
Regulation of ILC3s function and asthma
Although ILC3s are typically stimulated by IL-23 and IL-1β, they are also sensitive to environmental signals, such as caloric excess, micronutrients, and microbiota. A vitamin A deficit in mice resulted in greatly decreased numbers of ILC3s in the intestine, which increased the susceptibility of these mice to bacterial infections. Subsequently, treatment with vitamin A restored the number of ILC3s to normal levels; however, this treatment reduced the percentage of ILC2s [83]. In another study, vitamin D deficiency improved ILC3s responses (Fig. 2) [84]. ILCs are influenced by the ability of macrophages to sense microbial signals and produce IL-1β [85]. Interestingly, a study demonstrated that the AHR in obese mice was completely resolved with an IL-1 receptor antagonist, anakinra. The researchers also reported a decrease in the number of IL-17-producing lung ILC3s [72]. The microbiota possessed by asthmatic individuals in their airways is believed to have a higher potential to be pathogenic than that of non-asthmatic individuals [86]. How ILC3s contribute to and/or are impacted by the roles of these vitamins and the influence of this crosstalk with microbiota has not yet been evaluated.
Conclusion
Knowledge gained from recently recognized ILCs will help us to fill in the missing gaps of innate molecular pathways regarding asthma immunopathology. The lung ILCs on the frontier, sensitive to environmental factors including toxic and non-toxic substances, pathogenic and nonpathogenic microorganisms, and allergens, maintain homeostasis with tissue repair and remodeling. They can initiate AHR and appropriately set up the milieu for adaptive immunity by producing various cytokines, generally previously described in other contexts, and by interacting with different immune cells. ILCs represent one of the very first mediators for the different phenotypes of asthma ‘syndrome’ [10]. However, it is still unclear whether additional subsets of ILCs exist, and their role in innate immune memory has yet to be determined. We need further studies investigating their interaction with other immune cells, exogenous factors, and other micronutrients. A better understanding of their pathogenesis in asthma will be important for a better understanding of asthma phenotypes and for developing better strategies for preventive and therapeutic interventions.
References
Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy and Clinical Immunology. 2014. doi:10.1016/j.jaci.2014.11.001
Mjosberg J, Eidsmo L. Update on innate lymphoid cells in atopic and non-atopic inflammation in the airways and skin. Clin Exp Allergy. 2014;44(8):1033–43. doi:10.1111/cea.12353.
Mebius RE, Rennert P, Weissman IL. Developing lymph nodes collect CD4 + CD3- LTbeta + cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity. 1997;7(4):493–504.
Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. 2011;12(11):1045–54. doi:10.1031/ni.2131.
Artis D, Spits H. The biology of innate lymphoid cells. Nature. 2015;517(7534):293–301. doi:10.1038/nature14189.
Klose CS, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell. 2014;157(2):340–56. doi:10.1016/j.cell.2014.03.030.
Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, et al. T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med. 2014;211(3):563–77. doi:10.1084/jem.20131560.
Pulendran B, Artis D. New paradigms in type 2 immunity. Science. 2012;337(6093):431–5. doi:10.1126/science.1221064.
Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, et al. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326(5):298–304. doi:10.1056/nejm199201303260504.
Lotvall J, Akdis CA, Bacharier LB, Bjermer L, Casale TB, Custovic A, et al. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol. 2011;127(2):355–60. doi:10.1016/j.jaci.2010.11.037.
Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. 2012;18(5):716–25. doi:10.1038/nm.2678.
Agache I, Akdis C, Jutel M, Virchow JC. Untangling asthma phenotypes and endotypes. Allergy. 2012;67(7):835–46. doi:10.1111/j.1398-9995.2012.02832.x.
Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol. 2013;14(3):221–9. doi:10.1038/ni.2534.
Cella M, Miller H, Song C. Beyond NK cells: the expanding universe of innate lymphoid cells. Front Immunol. 2014;5:282. doi:10.3389/fimmu.2014.00282.
Korsgren M, Persson CG, Sundler F, Bjerke T, Hansson T, Chambers BJ, et al. Natural killer cells determine development of allergen-induced eosinophilic airway inflammation in mice. J Exp Med. 1999;189(3):553–62.
Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, et al. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med. 2013;5(174):174ra26. doi:10.1126/scitranslmed.3004812.
Huang YJ. The respiratory microbiome and innate immunity in asthma. Curr Opin Pulm Med. 2015;21(1):27–32. doi:10.1097/MCP.0000000000000124.
Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity. 2013;38(4):769–81. doi:10.1016/j.immuni.2013.02.010.
Saenz SA, Siracusa MC, Monticelli LA, Ziegler CG, Kim BS, Brestoff JR, et al. IL-25 simultaneously elicits distinct populations of innate lymphoid cells and multipotent progenitor type 2 (MPPtype2) cells. J Exp Med. 2013;210(9):1823–37. doi:10.1084/jem.20122332.
Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464(7293):1367–70. doi:10.1038/nature08900.
Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. 2010;463(7280):540–4. doi:10.1038/nature08636.
Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity. 2012;36(3):451–63. doi:10.1016/j.immuni.2011.12.020.
Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12(11):1055–62. doi:10.1038/ni.2104.
Bartemes KR, Kephart GM, Fox SJ, Kita H. Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J Allergy Clin Immunol. 2014;134(3):671–8. doi:10.1016/j.jaci.2014.06.024.
Allakhverdi Z, Comeau MR, Jessup HK, Yoon BR, Brewer A, Chartier S, et al. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J Exp Med. 2007;204(2):253–8. doi:10.1084/jem.20062211.
Angkasekwinai P, Park H, Wang YH, Wang YH, Chang SH, Corry DB, et al. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. 2007;204(7):1509–17. doi:10.1084/jem.20061675.
Oliphant CJ, Barlow JL, McKenzie AN. Insights into the initiation of type 2 immune responses. Immunology. 2011;134(4):378–85. doi:10.1111/j.1365-2567.2011.03499.x.
Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. 2008;20(6):791–800. doi:10.1093/intimm/dxn037.
Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage- CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. 2012;188(3):1503–13. doi:10.4049/jimmunol.1102832.
Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci U S A. 2010;107(25):11489–94. doi:10.1073/pnas.1003988107.
Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, et al. Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol. 2012;129(1):191–8 e1-4. 10.1016/j.jaci.2011.09.041.
Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, Broide DH. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J Allergy Clin Immunol. 2013;132(1):205–13. doi:10.1016/j.jaci.2013.03.048.
Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P, et al. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. J Allergy Clin Immunol. 2012;129(1):216–27 e1-6. doi:10.1016/j.jaci.2011.10.036.
Klein Wolterink RG, Kleinjan A, van Nimwegen M, Bergen I, de Bruijn M, Levani Y, et al. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur J Immunol. 2012;42(5):1106–16. doi:10.1002/eji.201142018.
Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, et al. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat Immunol. 2011;12(11):1071–7. doi:10.1038/ni.2133.
Bando JK, Nussbaum JC, Liang HE, Locksley RM. Type 2 innate lymphoid cells constitutively express arginase-I in the naive and inflamed lung. J Leukoc Biol. 2013;94(5):877–84. doi:10.1189/jlb.0213084.
Maarsingh H, Dekkers BG, Zuidhof AB, Bos IS, Menzen MH, Klein T, et al. Increased arginase activity contributes to airway remodelling in chronic allergic asthma. Eur Respir J. 2011;38(2):318–28. doi:10.1183/09031936.00057710.
Maarsingh H, Zaagsma J, Meurs H. Arginase: a key enzyme in the pathophysiology of allergic asthma opening novel therapeutic perspectives. Br J Pharmacol. 2009;158(3):652–64. doi:10.1111/j.1476-5381.2009.00374.x.
Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity. 2014;41(2):283–95. doi:10.1016/j.immuni.2014.06.016.
Drake LY, Iijima K, Kita H. Group 2 innate lymphoid cells and CD4+ T cells cooperate to mediate type 2 immune response in mice. Allergy. 2014;69(10):1300–7. doi:10.1111/all.12446.
Mirchandani AS, Besnard AG, Yip E, Scott C, Bain CC, Cerovic V, et al. Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J Immunol. 2014;192(5):2442–8. doi:10.4049/jimmunol.1300974.
Miranda C, Busacker A, Balzar S, Trudeau J, Wenzel SE. Distinguishing severe asthma phenotypes: role of age at onset and eosinophilic inflammation. J Allergy Clin Immunol. 2004;113(1):101–8. doi:10.1016/j.jaci.2003.10.041.
Miljkovic D, Bassiouni A, Cooksley C, Ou J, Hauben E, Wormald PJ, et al. Association between group 2 innate lymphoid cells enrichment, nasal polyps and allergy in chronic rhinosinusitis. Allergy. 2014;69(9):1154–61. doi:10.1111/all.12440.
Shaw JL, Fakhri S, Citardi MJ, Porter PC, Corry DB, Kheradmand F, et al. IL-33-responsive innate lymphoid cells are an important source of IL-13 in chronic rhinosinusitis with nasal polyps. Am J Respir Crit Care Med. 2013;188(4):432–9. doi:10.1164/rccm.201212-2227OC.
Ho J, Bailey M, Zaunders J, Mrad N, Sacks R, Sewell W, et al. Group 2 innate lymphoid cells (ILC2s) are increased in chronic rhinosinusitis with nasal polyps or eosinophilia. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2015;45(2):394–403. doi:10.1111/cea.12462.
Lam M, Hull L, McLachlan R, Snidvongs K, Chin D, Pratt E, et al. Clinical severity and epithelial endotypes in chronic rhinosinusitis. International forum of allergy & rhinology. 2013;3(2):121–8. doi:10.1002/alr.21082.
Mjosberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, et al. The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity. 2012;37(4):649–59. doi:10.1016/j.immuni.2012.08.015.
Singh AM, Moore PE, Gern JE, Lemanske Jr RF, Hartert TV. Bronchiolitis to asthma: a review and call for studies of gene-virus interactions in asthma causation. Am J Respir Crit Care Med. 2007;175(2):108–19. doi:10.1164/rccm.200603-435PP.
Bacharier LB, Boner A, Carlsen KH, Eigenmann PA, Frischer T, Gotz M, et al. Diagnosis and treatment of asthma in childhood: a PRACTALL consensus report. Allergy. 2008;63(1):5–34. doi:10.1111/j.1398-9995.2007.01586.x.
Gern JE. The ABCs of rhinoviruses, wheezing, and asthma. J Virol. 2010;84(15):7418–26. doi:10.1128/JVI.02290-09.
Jackson DJ, Evans MD, Gangnon RE, Tisler CJ, Pappas TE, Lee WM, et al. Evidence for a causal relationship between allergic sensitization and rhinovirus wheezing in early life. Am J Respir Crit Care Med. 2012;185(3):281–5. doi:10.1164/rccm.201104-0660OC.
Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12(7):631–8. doi:10.1038/ni.2045.
Gorski SA, Hahn YS, Braciale TJ. Group 2 innate lymphoid cell production of IL-5 is regulated by NKT cells during influenza virus infection. PLoS Pathog. 2013;9(9), e1003615. doi:10.1371/journal.ppat.1003615.
Hurst SD, Muchamuel T, Gorman DM, Gilbert JM, Clifford T, Kwan S, et al. New IL-17 family members promote Th1 or Th2 responses in the lung: in vivo function of the novel cytokine IL-25. J Immunol. 2002;169(1):443–53.
Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23(5):479–90. doi:10.1016/j.immuni.2005.09.015.
Gregory LG, Jones CP, Walker SA, Sawant D, Gowers KH, Campbell GA, et al. IL-25 drives remodelling in allergic airways disease induced by house dust mite. Thorax. 2013;68(1):82–90. doi:10.1136/thoraxjnl-2012-202003.
Ballantyne SJ, Barlow JL, Jolin HE, Nath P, Williams AS, Chung KF, et al. Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J Allergy Clin Immunol. 2007;120(6):1324–31. doi:10.1016/j.jaci.2007.07.051.
Cherry WB, Yoon J, Bartemes KR, Iijima K, Kita H. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J Allergy Clin Immunol. 2008;121(6):1484–90. doi:10.1016/j.jaci.2008.04.005.
Silver MR, Margulis A, Wood N, Goldman SJ, Kasaian M, Chaudhary D. IL-33 synergizes with IgE-dependent and IgE-independent agents to promote mast cell and basophil activation. Inflamm Res. 2010;59(3):207–18. doi:10.1007/s00011-009-0088-5.
Barlow JL, Peel S, Fox J, Panova V, Hardman CS, Camelo A, et al. IL-33 is more potent than IL-25 in provoking IL-13-producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J Allergy Clin Immunol. 2013;132(4):933–41. doi:10.1016/j.jaci.2013.05.012.
Liu X, Li M, Wu Y, Zhou Y, Zeng L, Huang T. Anti-IL-33 antibody treatment inhibits airway inflammation in a murine model of allergic asthma. Biochem Biophys Res Commun. 2009;386(1):181–5. doi:10.1016/j.bbrc.2009.06.008.
Levy BD, Serhan CN. Resolution of acute inflammation in the lung. Annu Rev Physiol. 2014;76:467–92. doi:10.1146/annurev-physiol-021113-170408.
Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8(5):349–61. doi:10.1038/nri2294.
Serhan CN, Yang R, Martinod K, Kasuga K, Pillai PS, Porter TF, et al. Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med. 2009;206(1):15–23. doi:10.1084/jem.20081880.
Krishnamoorthy N, Burkett PR, Dalli J, Abdulnour RE, Colas R, Ramon S, et al. Cutting edge: maresin-1 engages regulatory T cells to limit type 2 innate lymphoid cell activation and promote resolution of lung inflammation. J Immunol. 2015;194(3):863–7. doi:10.4049/jimmunol.1402534.
Xue L, Salimi M, Panse I, Mjosberg JM, McKenzie AN, Spits H, et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol. 2014;133(4):1184–94. doi:10.1016/j.jaci.2013.10.056.
Pettipher R, Hansel TT, Armer R. Antagonism of the prostaglandin D2 receptors DP1 and CRTH2 as an approach to treat allergic diseases. Nat Rev Drug Discov. 2007;6(4):313–25. doi:10.1038/nrd2266.
Lao-Araya M, Steveling E, Scadding GW, Durham SR, Shamji MH. Seasonal increases in peripheral innate lymphoid type 2 cells are inhibited by subcutaneous grass pollen immunotherapy. J Allergy Clin Immunol. 2014;134(5):1193–5. doi:10.1016/j.jaci.2014.07.029. 5.
Kudo M, Melton AC, Chen C, Engler MB, Huang KE, Ren X, et al. IL-17A produced by alphabeta T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat Med. 2012;18(4):547–54. doi:10.1038/nm.2684.
McKinley L, Alcorn JF, Peterson A, Dupont RB, Kapadia S, Logar A, et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol. 2008;181(6):4089–97.
Geremia A, Arancibia-Carcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med. 2011;208(6):1127–33. doi:10.1084/jem.20101712.
Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med. 2014;20(1):54–61. doi:10.1038/nm.3423.
Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature. 2013;498(7452):113–7. doi:10.1038/nature12240.
Taube C, Tertilt C, Gyulveszi G, Dehzad N, Kreymborg K, Schneeweiss K, et al. IL-22 is produced by innate lymphoid cells and limits inflammation in allergic airway disease. PLoS One. 2011;6(7):e21799. doi:10.1371/journal.pone.0021799.
Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464(7293):1371–5. doi:10.1038/nature08949.
Sun YC, Zhou QT, Yao WZ. Sputum interleukin-17 is increased and associated with airway neutrophilia in patients with severe asthma. Chin Med J (Engl). 2005;118(11):953–6.
Agache I, Ciobanu C, Agache C, Anghel M. Increased serum IL-17 is an independent risk factor for severe asthma. Respir Med. 2010;104(8):1131–7. doi:10.1016/j.rmed.2010.02.018.
Zhao Y, Yang J, Gao YD, Guo W. Th17 immunity in patients with allergic asthma. Int Arch Allergy Immunol. 2010;151(4):297–307. doi:10.1159/000250438.
Sherkat R, Yazdani R, Ganjalikhani Hakemi M, Homayouni V, Farahani R, Hosseini M, et al. Innate lymphoid cells and cytokines of the novel subtypes of helper T cells in asthma. Asia Pacific allergy. 2014;4(4):212–21. doi:10.5415/apallergy.2014.4.4.212.
Gibeon D, Batuwita K, Osmond M, Heaney LG, Brightling CE, Niven R, et al. Obesity-associated severe asthma represents a distinct clinical phenotype: analysis of the British Thoracic Society Difficult Asthma Registry Patient cohort according to BMI. Chest. 2013;143(2):406–14. doi:10.1378/chest.12-0872.
Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature. 2014. doi:10.1038/nature14115.
Lee MW, Odegaard JI, Mukundan L, Qiu Y, Molofsky AB, Nussbaum JC, et al. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell. 2015;160(1–2):74–87. doi:10.1016/j.cell.2014.12.011.
Spencer SP, Wilhelm C, Yang Q, Hall JA, Bouladoux N, Boyd A, et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science. 2014;343(6169):432–7. doi:10.1126/science.1247606.
Chen J, Waddell A, Lin YD, Cantorna MT. Dysbiosis caused by vitamin D receptor deficiency confers colonization resistance to Citrobacter rodentium through modulation of innate lymphoid cells. Mucosal immunology. 2014. doi:10.1038/mi.2014.94.
Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. 2014;343(6178):1249288. doi:10.1126/science.1249288.
Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5(1):e8578. doi:10.1371/journal.pone.0008578.
Acknowledgements
The authors wish to thank the European Academy of Allergy and Clinical Immunology, and its Junior Members and Affiliates for supporting the mentorship program and encouraging the authors to write this review. The authors also thank the artist, Merve Evren, for her work in creating the illustrations for this review.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LPO drafted the manuscript. HM and MA reviewed and finalized the manuscript. All authors read and approved the final manuscript.
An erratum to this article is available at http://dx.doi.org/10.1186/s13601-015-0075-6.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Ozyigit, L.P., Morita, H. & Akdis, M. Innate lymphocyte cells in asthma phenotypes. Clin Transl Allergy 5, 23 (2015). https://doi.org/10.1186/s13601-015-0068-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13601-015-0068-5