
Abstract
Sex differences are prevalent in normal development, physiology and disease pathogeneses. Recent studies have demonstrated that mosaic loss of Y chromosome and aberrant activation of its genes could modify the disease processes in male biased manners. This mini review discusses the nature of the genes on the human Y chromosome and identifies two general categories of genes: those sharing dosage-sensitivity functions with their X homologues and those with testis-specific expression and functions. Mosaic loss of the former disrupts the homeostasis important for the maintenance of health while aberrant activation of the latter promotes pathogenesis in non-gonadal tissues, thereby contributing to genetic predispositions to diseases in men.
Similar content being viewed by others


Sex differences and mosaic loss of Y chromosome
Sex differences are prevalent in development and physiology in humans, and pathogeneses of various diseases, such as cancers, neurodevelopmental, neurodegenerative and cardiovascular diseases [1,2,3,4,5,6,7,8]. Such sex differences in diseases include incidence, onset age, progression, phenotypes and treatment responses. Although sex hormones, i.e. androgens and estrogens, and their receptors, i.e. androgen and estrogen receptors, could play important roles in such biological processes [1, 3], genes on the male-specific Y chromosome could also contribute at the genetic levels to such differences between the sexes [9,10,11]. Recent studies on large populations with well-defined health information suggest that mosaic loss of the Y chromosome (mLOY) in the peripheral white blood cells predisposes men to various diseases, including cancers, cardiovascular and Alzheimer’s diseases [12,13,14,15,16,17], suggesting that the Y chromosome could be essential in maintaining the homeostasis important for the health of men [11]. Despite such associations, it is uncertain how mLOY in small portion of the leukocytes could contribute to the genetic predisposition to diseases in men. One hypothesis suggests that such mLOY in peripheral white blood cells is an indicator of genome instability in general, affecting genes in lymphocytes and other tissues and/or various physiological processes, such as inflammation, immunosurveillance and oxidative stresses [12, 13, 18], with functional consequences across diverse biological systems. Indeed, various novel and known loci in cell cycle regulation and cancer susceptibility have been associated with mLOY [12, 13, 19], thereby supporting the essential nature of the Y chromosome genes in the well-being of men. Mosaic LOY disrupts such balances and predisposes men to pathogenesis, resulting in sex differences in the various diseases.
Aberrant activation of Y chromosome genes potentiates pathogeneses of human diseases
Studies on individual Y chromosome genes have provided information suggesting that they could exert positive actions on the pathogeneses of various human diseases, including cancers and neurodevelopmental diseases, thereby preferentially affecting males [6, 20]. For example, expression of the sex-determining region Y (SRY) gene is elevated in dopamine neurons in human and experimental Parkinson’s disease models and suppression of its expression could exert protective functions against the disease in these models [21]. Studies on the spontaneously hypertensive rat (SHR) indicated that the rat Sry up regulates genes in the renin-angiotensin system, resulting in higher blood pressure in the male SHRs [22]. The human SRY could upregulate the monoamine oxidase A (MAOA) gene, whose expression levels are associated with various neurological and psychological disorders [23]. Further, aberrant expression of SRY could compete against the proper functions of a family of related transcription factors, encoded by the SRY-box (SOX) genes, which play critical roles in numerous developmental, physiological and pathogenic processes [24,25,26]. Indeed, SRY could impair the SOX10 regulation of the RET gene, important for enteric nervous system (ENS) differentiation [6]. Such impairment results in haploinsufficiency of the RET protein and exacerbation of the pathogenesis of the Hirschsprung’s disease, a congenital disorder affecting the ENS differentiation with significantly high male preference [6]. Importantly, aberrant activation of a human SRY transgene during embryogenesis in transgenic mice impairs the normal development of various vital organs, resulting in postnatal growth retardation and lethality [27]. Hence, these studies suggest that aberrant activation of Y chromosome genes, in this case SRY, could disrupt normal development and exacerbate the disease processes, thereby contributing to sex differences in a positive manner(s). These findings are in contrast to those of mLOY studies, in which loss of genes on the Y chromosome potentiates the disease processes [13, 15, 18, 19]. To understand the roles of the Y chromosome in human health and diseases, it is crucial to discuss a few basic aspects of the genetics and biological functions of the genes on this male-specific chromosome.
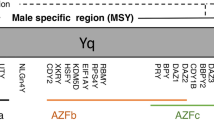
Genes on the human Y chromosome
In humans, men and women are genetically identical with 22 pairs of autosomes, except their sex chromosomes, i.e. X and Y chromosome. Men possess XY while women possess XX sex chromosome constitution. The gene dosage differences for the X chromosome is compensated by inactivation of the genes, with specific exceptions, on one of the X chromosomes during early development in females [28,29,30]. The mammalian X and Y chromosome originated from a pair of autosomes. One of them had acquired a male sex-determining gene, i.e. SRY, postulated to occur over 180 million years ago, and became the Y chromosome while the other homologue maintained the genetic content and structure of the ancestral chromosome and evolved to be the X chromosome [31]. Various evolutionary rearrangements and deterioration events resulted in a reduction in genetic content on the Y chromosome, which evolved to be the smallest and/or most gene-poor chromosome in most mammals, including humans [32]. The human Y chromosome is 57.23 MB in size and harbors two specific regions, generally referred to as the pseudoautosomal regions (i.e. PAR1 and PAR2) and male-specific region Y (MSY). PAR1 and PAR2 harbor ~ 2.6 MB and 320 kb of DNA and are located at the telomeric ends of the short and long arm of the Y chromosome respectively. The MSY is about 54 MB in size, consisting of approximately 24 MB of euchromatin and 30 MB of heterochromatin composed of mostly repetitive sequences (Genome Reference Consortium Human Build 38.p13). Cytogenetically, the heterochromatic long arm is highly variable, mostly involving either amplification or deletion of the heterochromatin and possibly the PAR2 at the telomere of the long arm [33, 34]. At present, there are 40 protein-coding genes/gene families on the human Y chromosome (Table 1) [35]. Fifteen genes are on the PAR1 and 3 genes are on the PAR2, which are also present in the PARs of the X chromosome. There is an obligatory crossover(s) between the X and Y chromosomes at their PARs during male meiosis [36], and hence genes on PARs behave similarly as those of the autosomes. The PAR genes escape X-inactivation and are mostly expressed in similar levels in both female and male tissues/cells [28] (Additional file 1: Figure S1A). Interestingly, there are 5 receptor genes, i.e. CRLF2, CSF2RA, IL3RA, P2RY8 and IL9R, among the PAR genes, involved in various cytokine and immune functions. Initial sequencing results suggested that there are 78 protein-coding genes on the MSY, corresponding to 27 distinct proteins [10]. This number varies due to microdeletions and copy number variations (CNV) of ampliconic genes among the general population [37]. There are 22 MSY genes, of which 17 are evolutionarily conserved with corresponding X homologues, widely expressed (Additional file 1: Figure S1B-C) and postulated to serve dosage-sensitive regulatory functions in chromatin modification, transcription, translation, RNA splicing and protein stability [9, 10], which likely exert global effects on gene expression and modification of protein functions. Gene ontology analysis of all 40 Y chromosome genes suggested that they could be associated with male fertility/infertility, autism, coronary and psychological/neurological diseases (Additional file 1: Table S1).
Noticeably, several MSY genes, such as SRY, TSPY and RBMY, are exceptions to such dosage-sensitive functions. They are primarily expressed in the testis and serve vital functions in the differentiation and physiology of this male-specific organ [38,39,40]. Their counterparts on the X chromosome are expressed ubiquitously in numerous tissue/cell types and mostly at similar levels between the sexes (Additional file 1: Figure S1D) and are subjected to X-inactivation [28]. Further, the respective encoded proteins could possess structural divergence(s), suggesting that they might serve different functions in various biological systems [24, 25, 41]. Accordingly, expression of these testis-specific MSY genes in somatic tissues could exert male-specific effects on the respectively affected organs/cells. We surmise that low-level/spatiotemporal expression of these MSY gene(s) during development/physiology could produce normal differences between the sexes [5, 42, 43], and aberrant/high level expression could result in male biases in the pathogeneses of various human diseases [6, 27, 41]. In particular, TSPY and its X homologue TSPX (TSPYL2) evolved from the same ancestral gene but diverged structurally in their encoded proteins to process contrasting functions in cell cycle regulation and androgen receptor (AR) transactivation [44, 45]. They represent a pair of homologues on the sex chromosomes, which could oppose each other in various biological processes [41]. TSPY is specifically expressed in the testis (Additional file 1: Figure S1E) and could serve important functions in spermatogonia stem cell renewal and male meiosis [46]. It is located on and is the putative gene for the gonadoblastoma locus on the Y chromosome (GBY) [47, 48]. It is frequently and aberrantly activated in various cancers, including gonadoblastoma, testicular germ cell tumors, melanoma, liver, head and neck and prostate cancers and promotes cell proliferation [48, 49]. TSPY interacts with AR and stimulates the AR transactivation of its target genes [45]. Importantly, TSPY is an androgen-responsive gene, and hence TSPY and AR form a positive feedback loop in amplifying their respective biological functions in male-specific manners [41]. TSPX is widely expressed equally in both male and female tissues (Additional file 1: Figure S1D) and is subjected to X-inactivation [50]. It retards cell proliferation and suppresses AR transactivation activities [45]. Accordingly, TSPY is a Y-located proto-oncogene and TSPX is an X-located tumor suppressor at the two extremes of the human oncogenic spectrum respectively [41]. This peculiar situation raises some very interesting scenarios on the roles of the X–Y homologues in cancers. As a proto-oncogene, abnormal activation of TSPY in somatic tissues could promote oncogenesis while an inactivation/deletion of TSPX could impair its tumor suppression function(s) [51, 52] specifically for males with only one X chromosome. If under certain conditions, TSPX escapes X-inactivation, it could increase the tumor suppression functions in females [29, 50]. Collectively, such aberrations could disproportionally exacerbate cancer initiation and progression in males.
Mosaic loss of the Y chromosome in 5–15% of the leukocytes of men represents one of the most common genetic abnormalities in humans [13,14,15,16, 18]. Since the Y chromosome includes the pseudoautosomal and male-specific regions, the loss of the PAR genes and those on MSY with dosage-sensitive functions could result in gene dosage deficiency in various biological systems. On the other hand, the testis-specific MSY genes are important for male-specific functions, and their loss might be less critical in such mLOY-mediated disease predispositions, except those associated with reproductive tissues. However, the abnormal expression of these-testis specific MSY genes in non-gonadal cells could modify the normal differentiation and physiology as well as the pathogenesis of the affected cells/tissues in male biased manners. Accordingly, there are two general categories of Y chromosome genes, those on the PARs and MSY with dosage-sensitivity and MSY genes with male/testis-specific expression and functions. Understanding the biology and genetics of individual genes on the Y chromosome could provide some clues on which genes are likely to be important for the maintenance of homeostasis and their losses or aberrant activations could contribute to disease predisposition in men.
Of mice and men
The laboratory mouse has been widely used as experimental models for human diseases using transgenic means [53, 54], including male and female animals with different sex chromosome constitutions [55, 56]. Beside sex determination, the genetic contents of the human and mouse Y chromosome are quite distinct [10, 57]. The mouse Y chromosome is ~ 92 Mb in size, likely one of the largest and most gene-rich Y chromosomes in mammals. Its PAR is ~ only 700 kb, located at the telomere of the long arm, and harbors only 3 genes, i.e. Asmt, Sts (pseudogene) and Mid1. The mouse MSY is > 90 Mb in size and is entirely euchromatic. It harbors ~ 700 genes, but only 8 ancestral genes, i.e. Sry, Zfy1/Zfy2, Usp9y, Ddx3y, Uty, Kdm5d, Tspy (pseudogene), and Rbmy, are conserved on the human Y chromosome (Table 1). Other orthologues of the human Y chromosome genes are located on either the X chromosome or autosomes of the mouse. The remainder ~ 690 genes of the mouse MSY are acquired repetitive genes, some of which are embedded in a 500-kb repeat unit, likely to be important for mouse fertility, but are not conserved in any mammalian Y chromosome [57]. Significantly, only two MSY genes, i.e. Sry (the sex-determining gene) and Eif2s3y (absent on the human Y chromosome), are sufficient to generate male mice with spermatogenesis [58]. Further, these two MSY genes could be replaced by the activation of Sox9 (the Sry downstream gene [26]) and Eif2s3x (the X-homologue of Eif2s3y) to produce similarly functional male mice without a Y chromosome [59]. Although transgenic mouse studies have provided critical information on the functional aspects on some of those conserved MSY genes [54, 58,59,60,61], studies on the other MSY genes of the human Y chromosome with the laboratory mouse might require some retrofitting of the mouse Y chromosome. In particular, the mouse Tspy is a recently evolved pseudogene, harboring numerous in-frame mutations disrupting its open reading frame. Transgenic studies had generated a specific line, TgTSPY9, which harbors ~ 50 copies of the an 8.2-kb human TSPY structural gene tandemly integrated on the Y chromosome of the mouse [62]. Since TSPY is tandemly repeated 30–60 times on the human Y chromosome [40], such tandem integration of the human transgene on the mouse Y chromosome resembles the organization of the human endogenous gene. Characterization of animals from this transgenic line showed that the human TSPY transgene is expressed in similar patterns as those of the human endogenous gene, including normal expression in spermatogonia and spermatocytes in the testis [62] and aberrant activation during oncogenesis [63]. Hence, TgTSPY9 is a humanized mouse model for TSPY gene. Accordingly, with the recent advances of genome editing and transgenic technologies [64,65,66,67], targeting the integration of human MSY gene(s) onto the mouse Y chromosome could be viable strategies to model and study their functions in biological processes, involved in various aspects of development, physiology and disease pathogeneses.
Availability of data and materials
Not applicable.
References
Regitz-Zagrosek V, Kararigas G. Mechanistic pathways of sex differences in cardiovascular disease. Physiol Rev. 2017;97(1):1–37.
Pike CJ. Sex and the development of Alzheimer’s disease. J Neurosci Res. 2017;95(1–2):671–80.
McEwen BS, Milner TA. Understanding the broad influence of sex hormones and sex differences in the brain. J Neurosci Res. 2017;95(1–2):24–39.
Zagni E, Simoni L, Colombo D. Sex and gender differences in central nervous system-related disorders. Neurosci J. 2016;2016:2827090.
Joyner MJ, Wallin BG, Charkoudian N. Sex differences and blood pressure regulation in humans. Exp Physiol. 2016;101(3):349–55.
Li Y, Kido T, Garcia-Barcelo MM, Tam PK, Tabatabai ZL, Lau YF. SRY interference of normal regulation of the RET gene suggests a potential role of the Y-chromosome gene in sexual dimorphism in Hirschsprung disease. Hum Mol Genet. 2015;24(3):685–97.
Gillies GE, Pienaar IS, Vohra S, Qamhawi Z. Sex differences in Parkinson’s disease. Front Neuroendocrinol. 2014;35(3):370–84.
Werling DM, Geschwind DH. Sex differences in autism spectrum disorders. Curr Opin Neurol. 2013;26(2):146–53.
Bellott DW, Hughes JF, Skaletsky H, Brown LG, Pyntikova T, Cho TJ, Koutseva N, Zaghlul S, Graves T, Rock S, et al. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature. 2014;508(7497):494–9.
Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, Repping S, Pyntikova T, Ali J, Bieri T, et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003;423(6942):825–37.
Maan AA, Eales J, Akbarov A, Rowland J, Xu X, Jobling MA, Charchar FJ, Tomaszewski M. The Y chromosome: a blueprint for men’s health? Eur J Hum Genet. 2017;25(11):1181–8.
Guo X, Dai X, Zhou T, Wang H, Ni J, Xue J, Wang X. Mosaic loss of human Y chromosome: what, how and why. Hum Genet. 2020;139(4):421–46.
Thompson DJ, Genovese G, Halvardson J, Ulirsch JC, Wright DJ, Terao C, Davidsson OB, Day FR, Sulem P, Jiang Y, et al. Genetic predisposition to mosaic Y chromosome loss in blood. Nature. 2019;575(7784):652–7.
Qin N, Li N, Wang C, Pu Z, Ma Z, Jin G, Zhu M, Dai M, Hu Z, Ma H, et al. Association of mosaic loss of chromosome Y with lung cancer risk and prognosis in a Chinese population. J Thorac Oncol. 2019;14(1):37–44.
Dumanski JP, Lambert JC, Rasi C, Giedraitis V, Davies H, Grenier-Boley B, Lindgren CM, Campion D, Dufouil C, European Alzheimer’s Disease Initiative I, et al. Mosaic Loss of Chromosome Y in Blood Is Associated with Alzheimer Disease. Am J Hum Genet. 2016;98(6):1208–19.
Forsberg LA, Rasi C, Malmqvist N, Davies H, Pasupulati S, Pakalapati G, Sandgren J, Diaz de Stahl T, Zaghlool A, Giedraitis V, et al. Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer. Nat Genet. 2014;46(6):624–8.
Haitjema S, Kofink D, van Setten J, van der Laan SW, Schoneveld AH, Eales J, Tomaszewski M, de Jager SCA, Pasterkamp G, Asselbergs FW, et al. Loss of Y chromosome in blood is associated with major cardiovascular events during follow-up in men after carotid endarterectomy. Circ Cardiovasc Genet. 2017;10(4):e001544.
Forsberg LA. Loss of chromosome Y (LOY) in blood cells is associated with increased risk for disease and mortality in aging men. Hum Genet. 2017;136(5):657–63.
Wright DJ, Day FR, Kerrison ND, Zink F, Cardona A, Sulem P, Thompson DJ, Sigurjonsdottir S, Gudbjartsson DF, Helgason A, et al. Genetic variants associated with mosaic Y chromosome loss highlight cell cycle genes and overlap with cancer susceptibility. Nat Genet. 2017;49(5):674–9.
Kido T, Lau YF. Roles of the Y chromosome genes in human cancers. Asian J Androl. 2015;17(3):373–80.
Lee J, Pinares-Garcia P, Loke H, Ham S, Vilain E, Harley VR. Sex-specific neuroprotection by inhibition of the Y-chromosome gene, SRY, in experimental Parkinson’s disease. Proc Natl Acad Sci USA. 2019;116(33):16577–82.
Milsted A, Underwood AC, Dunmire J, DelPuerto HL, Martins AS, Ely DL, Turner ME. Regulation of multiple renin-angiotensin system genes by Sry. J Hypertens. 2010;28(1):59–64.
Wu JB, Chen K, Li Y, Lau YF, Shih JC. Regulation of monoamine oxidase A by the SRY gene on the Y chromosome. Faseb J. 2009;23(11):4029–38.
Kamachi Y, Kondoh H. Sox proteins: regulators of cell fate specification and differentiation. Development. 2013;140(20):4129–44.
Sarkar A, Hochedlinger K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell. 2013;12(1):15–30.
Li Y, Zheng M, Lau YF. The sex-determining factors SRY and SOX9 regulate similar target genes and promote testis cord formation during testicular differentiation. Cell Rep. 2014;8(3):723–33.
Kido T, Sun Z, Lau YC. Aberrant activation of the human sex-determining gene in early embryonic development results in postnatal growth retardation and lethality in mice. Sci Rep. 2017;7(1):4113.
Patrat C, Ouimette JF, Rougeulle C. X chromosome inactivation in human development. Development. 2020;147(1):dev183095.
Wang D, Tang L, Wu Y, Fan C, Zhang S, Xiang B, Zhou M, Li X, Li Y, Li G, et al. Abnormal X chromosome inactivation and tumor development. Cell Mol Life Sci. 2020. https://doi.org/10.1007/s00018-020-03469-z.
Sierra I, Anguera MC. Enjoy the silence: X-chromosome inactivation diversity in somatic cells. Curr Opin Genet Dev. 2019;55:26–31.
Hughes JF, Page DC. The history of the Y chromosome in man. Nat Genet. 2016;48(6):588–9.
Hughes JF, Rozen S. Genomics and genetics of human and primate y chromosomes. Annu Rev Genomics Hum Genet. 2012;13:83–108.
Kuhl H, Rottger S, Heilbronner H, Enders H, Schempp W. Loss of the Y chromosomal PAR2-region in four familial cases of satellited Y chromosomes (Yqs). Chromosome Res. 2001;9(3):215–22.
Velissariou V, Sismani C, Christopoulou S, Kaminopetros P, Hatzaki A, Evangelidou P, Koumbaris G, Bartsocas CS, Stylianidou G, Skordis N, et al. Loss of the Y chromosome PAR2 region and additional rearrangements in two familial cases of satellited Y chromosomes: cytogenetic and molecular analysis. Eur J Med Genet. 2007;50(4):291–300.
O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733–45.
Burgoyne PS. Mammalian X and Y crossover. Nature. 1986;319(6051):258–9.
Jobling MA, Tyler-Smith C. Human Y-chromosome variation in the genome-sequencing era. Nat Rev Genet. 2017;18(8):485–97.
Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MJ, Foster JW, Frischauf AM, Lovell-Badge R, Goodfellow PN. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature. 1990;346(6281):240–4.
Zhang JS, Yang-Feng TL, Muller U, Mohandas TK, de Jong PJ, Lau YF. Molecular isolation and characterization of an expressed gene from the human Y chromosome. Hum Mol Genet. 1992;1(9):717–26.
Affara NA, Lau YF, Briggs H, Davey P, Jones MH, Khwaja O, Mitchell M, Sargent C. Report and abstracts of the first international workshop on Y chromosome mapping 1994. Cambridge, England, April 2–5, 1994. Cytogenet Cell Genet. 1994;67(4):359–402.
Lau YC, Li Y, Kido T. Battle of the sexes: contrasting roles of testis-specific protein Y-encoded (TSPY) and TSPX in human oncogenesis. Asian J Androl. 2019;21(3):260–9.
Cosgrove KP, Mazure CM, Staley JK. Evolving knowledge of sex differences in brain structure, function, and chemistry. Biol Psychiatry. 2007;62(8):847–55.
Paus T. Sex differences in the human brain: a developmental perspective. Prog Brain Res. 2010;186:13–28.
Li Y, Lau YF. TSPY and its X-encoded homologue interact with cyclin B but exert contrasting functions on cyclin-dependent kinase 1 activities. Oncogene. 2008;27(47):6141–50.
Li Y, Zhang DJ, Qiu Y, Kido T, Lau YC. The Y-located proto-oncogene TSPY exacerbates and its X-homologue TSPX inhibits transactivation functions of androgen receptor and its constitutively active variants. Hum Mol Genet. 2017;26(5):901–12.
Lau YF, Li Y, Kido T. Role of the Y-located putative gonadoblastoma gene in human spermatogenesis. Syst Biol Reprod Med. 2011;57(1–2):27–34.
Page DC. Hypothesis: a Y-chromosomal gene causes gonadoblastoma in dysgenetic gonads. Development. 1987;101:151–5.
Lau YF, Li Y, Kido T. Gonadoblastoma locus and the TSPY gene on the human Y chromosome. Birth Defects Res C Embryo Today. 2009;87(1):114–22.
Kido T, Lau YC. Identification of a TSPY co-expression network associated with DNA hypomethylation and tumor gene expression in somatic cancers. J Genet Genomics. 2016;43(10):577–85.
Li N, Carrel L. Escape from X chromosome inactivation is an intrinsic property of the Jarid1c locus. Proc Natl Acad Sci USA. 2008;105(44):17055–60.
Epping MT, Lunardi A, Nachmani D, Castillo-Martin M, Thin TH, Cordon-Cardo C, Pandolfi PP. TSPYL2 is an essential component of the REST/NRSF transcriptional complex for TGFbeta signaling activation. Cell Death Differ. 2015;22(8):1353–62.
Le Gallo M, O’Hara AJ, Rudd ML, Urick ME, Hansen NF, O’Neil NJ, Price JC, Zhang S, England BM, Godwin AK, et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet. 2012;44(12):1310–5.
Shpargel KB, Starmer J, Wang C, Ge K, Magnuson T. UTX-guided neural crest function underlies craniofacial features of Kabuki syndrome. Proc Natl Acad Sci USA. 2017;114(43):E9046–55.
Shpargel KB, Sengoku T, Yokoyama S, Magnuson T. UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet. 2012;8(9):e1002964.
Arnold AP. Conceptual frameworks and mouse models for studying sex differences in physiology and disease: why compensation changes the game. Exp Neurol. 2014;259:2–9.
Arnold AP, Chen X. What does the “four core genotypes” mouse model tell us about sex differences in the brain and other tissues? Front Neuroendocrinol. 2009;30(1):1–9.
Soh YQ, Alfoldi J, Pyntikova T, Brown LG, Graves T, Minx PJ, Fulton RS, Kremitzki C, Koutseva N, Mueller JL, et al. Sequencing the mouse Y chromosome reveals convergent gene acquisition and amplification on both sex chromosomes. Cell. 2014;159(4):800–13.
Yamauchi Y, Riel JM, Stoytcheva Z, Ward MA. Two Y genes can replace the entire Y chromosome for assisted reproduction in the mouse. Science. 2014;343(6166):69–72.
Yamauchi Y, Riel JM, Ruthig VA, Ortega EA, Mitchell MJ, Ward MA. Two genes substitute for the mouse Y chromosome for spermatogenesis and reproduction. Science. 2016;351(6272):514–6.
Yamauchi Y, Riel JM, Ruthig V, Ward MA. Mouse Y-encoded transcription factor Zfy2 is essential for sperm formation and function in assisted fertilization. PLoS Genet. 2015;11(12):e1005476.
Koopman P, Gubbay J, Vivian N, Goodfellow P, Lovell-Badge R. Male development of chromosomally female mice transgenic for Sry. Nature. 1991;351(6322):117–21.
Schubert S, Skawran B, Dechend F, Nayernia K, Meinhardt A, Nanda I, Schmid M, Engel W, Schmidtke J. Generation and characterization of a transgenic mouse with a functional human TSPY. Biol Reprod. 2003;69(3):968–75.
Kido T, Schubert S, Hatakeyama S, Ohyama C, Schmidtke J, Lau YF. Expression of a Y-located human proto-oncogene TSPY in a transgenic mouse model of prostate cancer. Cell Biosci. 2014;4(1):9.
Zhu F, Nair RR, Fisher EMC, Cunningham TJ. Humanising the mouse genome piece by piece. Nat Commun. 2019;10(1):1845.
Heidenreich M, Zhang F. Applications of CRISPR-Cas systems in neuroscience. Nat Rev Neurosci. 2016;17(1):36–44.
Uno N, Abe S, Oshimura M, Kazuki Y. Combinations of chromosome transfer and genome editing for the development of cell/animal models of human disease and humanized animal models. J Hum Genet. 2018;63(2):145–56.
Zhao X, Wei W, Pan H, Nie J, Chen D, Zhang P, Chen F, Fu Q, Zuo E, Lu Y, et al. Identification of the sex of pre-implantation mouse embryos using a marked Y chromosome and CRISPR/Cas9. Sci Rep. 2019;9(1):14315.
Funding
This work is partially supported by a grant from the Department of Defense Peer-Reviewed Cancer Program to YFCL (CA150248) and a Merit-Review Grant IO1 BX004446 from the Department of Veterans Affairs. Y-FC Lau is the recipient of a Research Career Scientist award (1 IK6BX004854) from the Department of Veterans Affairs.
Author information
Authors and Affiliations
Contributions
The author conceived and wrote the paper. The author read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The Author has written and consented for the publication of this manuscript.
Competing interests
The author declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Examples of expression patterns of Y chromosome genes in 54 normal male (blue) and female (red) human tissues in the Genotype-Tissue Expression (GTEx) Project database, analyzed on 05/13/2020.
Additional file 2: Table S1.
Diseases and pathways associated with Y chromosome genes revealed by gene ontology analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lau, YF.C. Y chromosome in health and diseases. Cell Biosci 10, 97 (2020). https://doi.org/10.1186/s13578-020-00452-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-020-00452-w