
Abstract
Introduction
[18F]MC225 is a selective substrate for P-glycoprotein (P-gp) that has good metabolic stability and shows higher baseline uptake compared with other P-gp substrates such as (R)-[11C]Verapamil. Prior to clinical translation, it is necessary to perform process validation of the radiosynthesis, assessment of preclinical toxicity, and radiation dosimetry.
Methods
The production of [18F]MC225 was automated on a CFN-MPS200 multipurpose synthesizer. The acute toxicity of MC225 was evaluated at a dose of 2.5 mg/kg bodyweight, which is more than 10,000-fold the postulated maximum clinical dose of [18F]MC225. The acute toxicity of [18F]MC225 injection at a 200-fold dose, to administer a postulated dose of 185 MBq of [18F]MC225, was also evaluated after the decay-out of 18F. The mutagenicity of MC225 was studied by a reverse mutation test using Salmonella typhimurium and Escherichia coli (Ames test). In vivo biodistribution and dosimetry studies of [18F]MC225 were carried out in normal mice. Human dosimetry was estimated using OLINDA software.
Results
The mean decay-corrected yields of [18F]MC225 at end of synthesis were 13%, with > 99% radiochemical purity, > 1000 GBq/μmol molar activity, and ≤ 1.5 μg/185 MBq of total chemical contents. All process validation batches complied with the product specifications and the process was confirmed to be appropriate for the production of [18F]MC225. No acute toxicity of MC225 or [18F]MC225 injection was found. No mutagenic activity was observed for MC225. The biodistribution study demonstrated both hepatobiliary and renal excretion of radioactivity. The most critical organ was the pancreas, with (63.8 μGy/MBq) or without urination (63.9 μGy/MBq) at 360 min after injection. The estimated effective dose (μSv/MBq) with and without urination at 360 min after injection was calculated as 15.7 and 16.9, respectively.
Conclusion
[18F]MC225 shows acceptable pharmacological safety at the dose required for adequate PET imaging. The potential risk associated with [18F]MC225 PET imaging is well within acceptable dose limits.
Similar content being viewed by others

Introduction
The blood-brain barrier (BBB) plays an important role in protecting the brain from xenobiotics and in maintaining homeostasis in the internal environment of the central nervous system (CNS) [1]. P-glycoprotein (P-gp) is an ATP-binding cassette transporter that is constitutively expressed in the luminal membrane of the BBB. It protects brain tissue against small hydrophobic xenobiotics that can passively diffuse through the BBB by selectively transporting them from cells into the extracellular space [2]. Hence, P-gp may also limit or prevent access of drugs such as antiepileptics, antidepressants, and anticancer agents to their target site in the brain [3]. Multiple clinical and preclinical evidence suggests that enhanced P-gp function at the BBB may be responsible for drug resistance in several diseases, including epilepsy [4,5,6,7,8], depression [9, 10], and human immunodeficiency virus infection and acquired immune deficiency syndrome [11,12,13]. Furthermore, altered P-gp function at the BBB has been proposed as a possible etiology of neurodegenerative disease; for example, decreased P-gp function may decrease clearance of β-amyloid from interstitial fluid in the brain to the plasma, which would result in a predisposition for β-amyloid deposition in Alzheimer’s disease [14,15,16,17,18,19,20]. A significant decrease of P-gp function in Parkinson’s disease patients is likely to facilitate the accumulation of toxic compounds in the brain [21,22,23].
Several potent P-gp substrates, including (R)-Verapamil, have been labeled for imaging P-gp function with positron emission tomography (PET) [24, 25]. These substrates have high affinity for P-gp and measure decreased function as increased tracer uptake by the brain. However, they are not likely to measure overexpression of P-gp because the concentration of the tracer is already almost unmeasurable at baseline [24,25,26,27]. Besides low brain uptake of (R)-[11C]verapamil, another disadvantage of this radiotracer is the formation of labeled metabolites, which also act as P-gp substrates [28].
5-(1-(2-[18F]fluoroethoxy))-[3-(6,7-dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)-propyl]-5,6,7,8-tetrahydronaphthalen ([18F]MC225) has recently been developed as a selective substrate for P-gp with good metabolic stability and has shown higher baseline uptake than that of other P-gp substrates [29, 30] because it is a weak substrate. These are suitable properties for measuring overexpression of P-gp in the brain. Preclinical studies have proved that [18F]MC225 has sufficient sensitivity to detect daily fluctuation of P-gp function in the rodent brain [31]. Very recently, a head-to-head comparison of [18F]MC225 with (R)-[11C]Verapamil in non-human primates showed a higher baseline uptake of [18F]MC225, which makes it a suitable tracer for measuring overexpression of P-gp [32]. These findings prompted us to undertake initial evaluation of [18F]MC225 in human subjects as a phase 1 study. As the first step prior to clinical application in humans, we performed a process validation of [18F]MC225 radiosynthesis for clinical use and assessed the preclinical toxicity and radiation dosimetry estimated from mouse distribution data.
Parts of this study have been published as a poster in 2019 [33].
Materials and methods
General
MC225 and its phenol precursor, 5-(1-(2-hydroxy))-[3-(6,7-dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)-propyl]-5,6,7,8-tetrahydronaphthalen (MC226) was custom synthesized by Nard Institute (Kobe, Japan) according to the methods described previously [29]. 2-Bromoethyl tosylate was purchased from ABX (Radeberg, Germany). All other chemical reagents were obtained from commercial sources. Male ddY mice were obtained from Japan SLC (Shizuoka, Japan). Sprague-Dawley [Crl:CD(SD)] rats were obtained from Charles River Laboratories (Atsugi, Japan). The animal studies were approved by the Animal Care and Use committee of the Tokyo Metropolitan Institute of Gerontology (Approval Nos. 17079, 17080, and 18016) and BoZo Research Center (Approval Nos. K180026 and K180027). Acute toxicity studies were performed under the guidelines of the “Revision of Guidelines for Single-Dose and Repeated-Dose Toxicity Studies”, Notification No. 88 of the Pharmaceutical Affairs Bureau, Ministry of Health and Welfare, Japan, August 10, 1993. Ames test was performed under the “Guidelines for Genotoxicity Studies of Pharmaceuticals” Yakushoku-shinsa No. 0920-1: September 20, 2012, Pharmaceutical and Food Safety Bureau, Ministry of Health, Labour and Welfare (MHLW).
Automated synthesis
Setup of automated synthesizer
[18F]MC225 was produced on a CFN-MPS200 multipurpose synthesizer (Sumitomo Heavy Industries, Tokyo, Japan) with a custom-made disposable cassette and an integrated high-performance liquid chromatography (HPLC) purification unit and solid-phase extraction (SPE) formulation unit with a sterile disposable cassette. Figure 1 shows the cassette setup, including tubing connections, vials, and other disposables. The custom-made cassettes were pre-assembled in a cleanroom using disposable materials supplied by Sumitomo Heavy Industries. We selected PharMed® BOT tube (Saint-Gobain, Akron, OH, USA) because of its good general chemical resistance and excellent acid, alkali, and oxidation resistance. Table 1 lists the module setup of reagents in detail. A Sep-Pak Accell Plus QMA Light Cartridge (Waters, Milford, MA, USA) preconditioned with 10 mL 1 M K2CO3 solution followed by 60 mL water for injection (Otsuka Pharmaceutical Factory, Naruto, Japan) and a Sep-Pak tC18 Plus Short Cartridge (Waters) preconditioned with 5 mL EtOH followed by 40 mL water for injection (Otsuka Pharmaceutical Factory) were installed between the VP34–VP35 and the VH17–VH18 positions, respectively.

Setup of the synthesis cassette, reagents, and vials for [18F]MC225 production with integrated HPLC separation and SPE formulation units
Process description
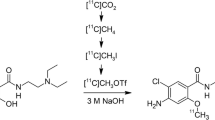
[18F]MC225 was synthesized as previously reported by Savolainen et al. [29] via a two-pot reaction (Fig. 2). No-carrier-added 18F-fluoride was produced by bombardment of [18O]H2O (Taiyo Nippon Sanso, Tokyo, Japan) at 50 μA × 20 min with 20 MeV protons on a HM-20 cyclotron system (Sumitomo Heavy Industries). The target water containing 18F-fluoride was transferred directly to the V-vial. The irradiated [18O]H2O was passed through the QMA-cartridge connected between VP34 and VP35, and the enriched water was recovered in an 18O-water recovery vial. The QMA-trapped 18F-fluoride was eluted with a reagent of R1 [mixture of aqueous 0.5 mL of K2CO3 (20 μmol) and a 2.0 mL MeCN solution of K.222 (40 μmol)] into the RV1. The solvents were removed azeotropically with MeCN at 100 °C under a slight flow of N2 and vacuum over 15 min. This procedure was conducted once more with 1.0 mL MeCN from R2. The RV1 was then cooled to 40 °C prior to the next step. The reagent of R3 [20 mg of 2-bromoethyl tosylate (61 μmol) in 1.0 mL of o-DCB] was transferred to the RV1, and distillation of the formed 2-bromoethyl [18F]fluoride was started immediately at 90 °C with N2 gas flow to RV2 containing 2 mg of MC226 (5.2 μmol) and 3 mg of NaH (125 μmol) in 0.5 mL of DMF at room temperature. After radioactivity of RV2 reached a plateau (~ 7 min), RV2 was reacted for 5 min at 80 °C. After cooling to 40 °C, 2.0 mL of quenching solution in R4 was added and the reaction mixture was transferred to the reservoir of the HPLC separation unit. The product was separated by HPLC [column: Agilent Eclipse XDB-C18 (5 μm, 250 mm × 9.4 mm inner diameter; solvent: MeCN/50 mM AcONH4 = 65/35], at a flow rate of 5 mL/min. The eluent was monitored by UV 254 nm, and radioactivity detectors were connected in series. The fraction of [18F]MC225 (retention time = 9 min) was collected into dilution bottle sR3, which was preloaded with 100 mL water for injection (Otsuka Pharmaceutical Factory) containing 1 mL of 250 mg/mL ascorbate injection (Nipro Pharma, Osaka Japan). After mixing with N2, the solution was transferred to a tC18 cartridge connected between the VH17–VH18 positions. [18F]MC225 was trapped in the cartridge, which was washed with 10 mL of water for injection (sR1), and the product was eluted with 1.4 mL of EtOH (sR2) into a Formulation Vial preloaded with 15 mL of formulation buffer solution [ascorbate injection/polysorbate 80 (Fujifilm Wako Pure Chemical, Osaka Japan)/sterile saline (Otsuka Pharmaceutical Factory) = 0.5/0.1/20]. After gentle mixing with N2, the solution was transferred and passed through a 0.22-μm sterilizing filter (Millex GV; Merck Millipore, Darmstadt, Germany) into an empty 30 mL sterile vial (Mita Rika Kogyo, Osaka, Japan) fitted with a sterile-filtered venting needle (Terumo, Tokyo, Japan).

Radiosynthesis of [18F]MC225
Quality control
Filter integrity was assessed by bubble point test (SLTEST000; Merck Millipore). The pH value of the injection solution was determined using a pH meter (LAQUA F-72; Horiba, Kyoto, Japan). Residual solvents in the injection solution were measured by capillary gas chromatography using a GC-2014 system with LabSolutions Software (Shimadzu, Kyoto, Japan). The radionuclide was identified by determining the half-life measurement using a Capintec CRC-55tR dose calibrator (Florham Park, NJ, USA) and the measurement of the 511 keV photopeak using a NaI(Tl) radiation detector (US-2000; Universal Giken, Odawara, Japan). Residual K.222 in the injection solution was evaluated by spot test. The limulus amebocyte lysate (LAL) test was conducted on a Toxinometer® ET-6000 (Fujifilm Wako Pure Chemical). Finally, a sample of the product formulation was tested for sterility post-release using direct inoculation in accordance with the Japanese Pharmacopoeia, 17th edition.
HPLC analysis was performed on a Shimadzu Prominence HPLC system equipped with a model LC-20AD pump, model SPD-20A UV absorbance detector (set at 280 nm), a GABI 3 × 3 in. NaI scintillation detector (Elysia-Raytest, Straubenhardt, Germany), and an analytical column (YMC-Pack ODS-A, 3 μm, 50 mm × 2.1 mm inner diameter) purchased from YMC (Kyoto, Japan). Operation of the Shimadzu Prominence HPLC system was controlled using Shimadzu LabSolutions software. For analysis, isocratic elution was applied using MeCN/50 mM AcONH4 solution = 60/40 (flow rate = 1 mL/min). Retention time of the authentic standard MC225 was 7.6 min.
Acute toxicity
Toxicity studies of MC225 were performed at the Kannami Laboratory, BoZo Research Center (Shizuoka, Japan). Acute toxicity was assayed in Sprague-Dawley rats [Crl:CD(SD)]. MC225 at a dose of 2.5 mg/kg bodyweight (0.5 mg/mL in 10 w/v% DMSO containing water for injection) was injected intraperitoneally into 6-week-old rats weighing 226–236 g (males, n = 5) and 149–161 g (females, n = 5). The dose of 2.5 mg/kg bodyweight is the 10,000-fold equivalent of the postulated maximum administration dose (0.25 μg/kg bodyweight) of 370 MBq [18F]MC225, with the lowest molar activity of 6.3 MBq/nmol for humans weighing 40 kg. Rats were observed frequently until 30 min and then at 1, 2, 4, and 6 h after the injection on day 1, and thereafter once daily for 14 days for clinical signs of toxicity. Rats were weighed on days 1, 2, 4, 7, and 14. At the end of the 14-day observation period, the rats were euthanized by exsanguination under isoflurane anesthesia, and a macroscopic analysis of the autopsy samples was performed. Three batches of [18F]MC225 were prepared and assayed after the decay-out of 18F. Solutions with decayed [18F]MC225 were individually injected intravenously into 6-week-old male and female rats (n = 3 each) at doses of 13.15 μg/5.29 mL/kg bodyweight, 9.22 μg/4.63 mL/kg bodyweight, and 1.68 μg/3.91 mL/kg bodyweight, for each of the three batches, equivalent to 200-fold of the postulated administration dose of 185 MBq [18F]MC225 for humans. After injection of decayed [18F]MC225, the rats were observed for clinical signs of toxicity for 14 days, and a macroscopic analysis was then performed as described above.
Mutagenicity
Mutagenicity tests were performed at the Tokyo Laboratory, BoZo Research Center (Tokyo, Japan). MC225 was tested for mutagenicity by the Ames test with four histidine-requiring strains of Salmonella typhimurium (TA98, TA100, TA1535, and TA1537) and one strain of Escherichia coli (WP2uvrA), with and without the S9 mixture, at a dose range of 19.5–5000 μg/plate according to the standard method.
Dosimetry
[18F]MC225 (2.1 MBq/1.6 pmol) was injected intravenously into 8-week-old male ddY mice. The tracer-injected mice were housed individually in filter-paper-lined animal-rearing cages until the time of euthanasia. Mice were killed by cervical dislocation at 5, 15, 30, 60, 180, and 360 min after injection (n = 4 each). The blood was collected by heart puncture, and the tissues were harvested. Radioactivity excreted into the urine was recovered from the cage floor and by cystocentesis from the urinary bladder. The samples were measured for 18F radioactivity with an auto-gamma counter (Hidex-AMG, Turk, Finland) and weighed. The tissue uptake of 18F was expressed as the percentage of injected dose per organ (%ID/organ) or the percentage of injected dose per gram of tissue (%ID/g). The tissue distribution data were extrapolated to an adult male phantom using the %kg/g method [34]. The radiation absorbed dose and effective dose for human adults were estimated using OLINDA/EXM software (Vanderbilt University, Nashville, TN, USA) [35].
Results
Automated synthesis
The three production runs had activity yields of 3527 ± 965 MBq, decay-corrected yields of 12.8 ± 2.6%, molar activity of 1576 ± 446 GBq/μmol, and radiochemical purity of 99.5 ± 0.3% (Table 2). The average synthesis time following target bombardment was 74 min. All batches of [18F]MC225 injection met the QC criteria listed in Table 2. [18F]MC225 was stable for up to 2 h after end of synthesis, with acceptable appearance, pH of 6.2 ± 0.1, and radiochemical purity of 98.3 ± 0.3%. Figure 3 shows a representative semi-preparative HPLC chromatogram of the reaction mixture. The phenol precursor (MC226), which elutes at approximately 6 min, was well separated from the product (MC225), which elutes at approximately 9 min.

Representative preparative HPLC chromatogram of the reaction mixture
Acute toxicity
Acute toxicity in rats was evaluated after a single intraperitoneal injection of MC225 at a dose of 2.5 mg/kg and a single intravenous injection of one of the three lots of [18F]MC225 preparations at a dose range of 1.68–13.15 μg/kg. There was no mortality in the rats during the 14-day observation period. All rat groups showed normal gains in bodyweight compared with the control animals, and no clinical signs of toxicity were observed over the 15-day period. Postmortem macroscopic examination found no abnormalities.
Mutagenicity
A bacterial reverse mutation test conducted using Salmonella thyphimurium and Escherichia coli detected no mutagenic activity for MC225.
Dosimetry
The tissue distribution of radioactivity after injection of [18F]MC225 into mice is summarized in Fig. 4, and in Tables 3 and 4. The radioactivity concentrations in the blood decreased rapidly after [18F]MC225 injection. The lung and kidney showed initial high uptake (%ID/g) before decreasing gradually (Fig. 4a). Among all of the examined organs, the pancreas showed the highest radioactivity concentration, reaching 37%ID/g at 180 min, which was maintained at 33%ID/g for 360 min after injection (Fig. 4a). Excretion of radioactivity into the bladder and urine increased gradually in response to the clearance of radioactivity from the kidney, reaching 18%ID/organ (bladder + urine) at 360 min after injection (Fig. 4b). Radioactivity of the liver peaked (24%ID/organ) at 15 min after injection, before clearing (Fig. 4b). In response to the clearance of radioactivity from the liver, radioactivity levels of the small intestine peaked at 180 min (22%ID/organ), and radioactivity of the large intestine gradually increased to reach 20%ID/organ at 360 min after injection (Fig. 4b). These data demonstrate that radioactivity was excreted by both the hepatobiliary and renal urinary systems. The radiation absorbed dose was estimated from these biodistribution data (Table 5). The absorbed dose (μGy/MBq) calculated with urination at 360 min after injection was highest in the pancreas (63.8), small intestine (36.0), urinary bladder wall (32.5), and upper large intestine wall (30.4). The absorbed dose calculated without urination was highest in the pancreas (63.9), urinary bladder wall (59.6), small intestine (36.3), and upper large intestine wall (30.6). The effective dose according to the risk-weighting factors of ICRP103 [36] was estimated as 15.7 μSv/MBq (with urination at 360 min after injection) and 16.9 μSv/MBq (without urination).

Regional decay-corrected time activity curves showing high radioactivity concentration (a) and accumulation (b) after intravenous injection of [18F]MC225 into mice. Data are presented as the mean ± S.D. (n = 4)
Discussion
We automated the radiolabeling process of [18F]MC225 on a CFN-MPS200 multipurpose synthesizer. The obtained activity yields (2650–4561 MBq) and high molar activity (1112–2002 GBq/μmol) were good enough for clinical research purposes. We started the radiosynthesis from approximately 40 GBq of 18F-fluoride (QMA-trapped radioactivity). It is possible to further improve activity yields and molar activity by starting with higher levels of 18F-fluoride radioactivity. The high molar activity of [18F]MC225 probably resulted from using fluoride-free materials (e.g., silicon tubes, thermoplastic elastomer tubes, polypropylene connectors) in assembly of the cassette of the CFN-MPS200. All three validation runs of [18F]MC225 satisfied the QC release criteria. No degradation of [18F]MC225 in formulation was confirmed up to 2 h. Taken together, the radiolabeling process of [18F]MC225 on a CFN-MPS200 was considered to be of sufficient quality for clinical use. This synthesis process has been approved for human use by the Institutional PET Drug Committee and MHLW Certified Clinical Research Review Board, Tokyo Metropolitan Geriatric Medical Center (CRB3180026) and is now in use for first-in-human clinical research (jRCTs031190136).
The absence of any abnormality in rats in the acute toxicity test together with the absence of mutagenicity of MC225 demonstrated the clinical suitability of [18F]MC225 for PET studies in humans. The estimated NOAEL of MC225 is > 5.85 μmol/2.5 mg/kg, which is the 10,000–fold equivalent of the postulated maximum administration dose. Because PET tracers are administered intravenously as a bolus, it is important to evaluate their potential toxic hazard to the cardiovascular system. Fusi et al. evaluated the effect of MC225 on the mechanical activity of freshly isolated rat aortic rings and on Cav1.2 channel current (ICa1.2) of A7r5 cells, as well as on cardiac function and electrocardiogram in Langendorff-perfused isolated rat hearts [37]. These in vitro cardiovascular studies demonstrated that MC225 has potential to induce vasodilator action and cardiotoxic effect over the 1000-fold equivalent postulated maximum administration dose (0.58 nmol/0.25 μg/kg) of MC225. All three validation runs demonstrated high molar activity of [18F]MC225 (> 1000 GBq/μmol) and a low amount of MC225-related chemical impurities. Because the estimated total chemical contents are ≤ 1.5 μg/185 MBq, the potential risk associated with [18F]MC225 injection is considered to be within the toxicologically acceptable range [38].
The radiation absorbed dose was highest in the pancreas, followed by the urinary bladder, small intestine wall, and large intestine wall. Urination at 360 min after injection significantly decreased the absorbed dose in the urinary bladder. Except for the pancreas, all of the organs with high absorbed dose are in the excretion route. The effective dose was well within the previously reported range (15–30 μSv/MBq) for 18F-labeled PET radiopharmaceuticals [39]. In the case of administration of 185 MBq of [18F]MC225, effective dose is estimated as 2.9 mSv (with urination at 360 min after injection) and 3.1 mSv (without urination), which is within the strict limit of 10 mSv set by the ICRP recommendations [40] and practiced in Europe. In this condition, the highest absorbed dose in the pancreas (with or without urination) is estimated as 11.8 mGy, which is also within the strict limits for individual organs (30 mSv for sensitive organs and 50 mSv for all others), as required by US Radioactive Drug Research Committee regulations [41].
There have been several reports indicating that preclinical (i.e., animal-derived) dosimetry of 18F-labeled tracers underestimates 20–40% of the effective dose to humans [42]. Taking into account the discrepancy between extrapolated and actually determined data in humans, we calculated effective dose of [18F]MC225 dividing by 0.6 to correct 40% underestimation to humans. The corrected dose (26.1 and 28.1 μSv/MBq for urination and without urination, respectively) was still within the previously reported ranges of 18F-labeled PET radiopharmaceuticals [39]. Furthermore, administration of 185 MBq of [18F]MC225 was still within the strict limit of 10 mSv (4.8 and 5.2 mSv for urination and without urination, respectively).
Conclusion
The automated synthesis of [18F]MC225 for clinical use was successfully and efficiently achieved on a CFN-MPS200 multipurpose synthesizer. All three process validation batches complied with the product specifications, and the process was confirmed to be appropriate for the production of [18F]MC225. Preclinical toxicological studies indicated that [18F]MC225 shows acceptable pharmacological safety at the dose required for adequate PET imaging. Taking into account the high molar activity and low total chemical contents, [18F]MC225 will be tolerable in first-in-human clinical trials. A single intravenous injection of 185 MBq of [18F]MC225 leads to an estimated effective dose of 2.9 mSv (with urination at 360 min after injection) and 3.1 mSv (without urination), and highest absorbed dose to an organ of 11.8 mGy (with or without urination). The potential risk associated with [18F]MC225 PET imaging is well within acceptable dose limits.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AcONH4 :
-
Ammonium acetate
- BBB:
-
Blood-brain barrier
- CNS:
-
Central nervous system
- o-DCB:
-
o-Dichlorobenzene
- DMF:
-
N, N-Dimethylformamide
- DMSO:
-
Dimethyl sulfoxide
- EtOH:
-
Ethanol
- EU:
-
Endotoxin unit
- HPLC:
-
High performance liquid chromatography
- ICRP:
-
International Commission on Radiological Protection
- K.222:
-
4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane
- LAL:
-
Limulus amebocyte lysate
- MeCN:
-
Acetonitrile
- MC225:
-
5-(1-(2-fluoroethoxy))-[3-(6,7-dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)-propyl]-5,6,7,8-tetrahydronaphthalen
- MC226:
-
5-(1-(2-hydroxy))-[3-(6,7-dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)-propyl]-5,6,7,8-tetrahydronaphthalen
- MHLW:
-
Ministry of Health, Labor and Welfare
- N/A:
-
Not applicable
- N.D:
-
Not detected
- NOAEL:
-
No observable adverse effect level
- %ID/g:
-
Percentage of injected dose per gram of tissue
- %ID/organ:
-
Percentage of injected dose per organ
- PET:
-
Positron emission tomography
- P-gp:
-
P-glycoprotein
- QC:
-
Quality control
- Verapamil:
-
2-(3,4-dimethoxyphenyl)-5-[2-(3,4-dimethoxyphenyl)ethyl-methylamino]-2-propan-2-ylpentanenitrile
- SPE:
-
Solid phase extraction
- S.D.:
-
standard deviation
- UV:
-
Ultraviolet
References
Terasaki T, Ohtsuki S. Brain-to-blood transporters for endogenous substrates and xenobiotics at the blood-brain barrier: an overview of biology, and methodology. NeuroRx. 2005;2:63–72.
Giacomini KM, Huang S-M. Transporters in drug development and clinical pharmacology. Clin Pharmacol Ther. 2013;94:3–9.
Graff CL, Pollack GM. Drug transport at the blood-brain barrier and the choroid plexus. Curr Drug Metab. 2004;5:95–108.
Löscher W, Postchka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci. 2005;6:591–602.
Feldmann M, Koepp M. ABC transporters and drug resistance in patients with epilepsy. Curr Pharm Des. 2016;22:5793–807.
Bauer M, Karch R, Zeitlinger M, Liu J, Koepp MJ, Asselin M-C, Sisodiya SM, Hainfellner JA, Wasdak W, Mitterhauser M, Müller M, Pataraia E, Langer O. In vivo P-glycoprotein function before and after epilepsy surgery. Neurology. 2014;83:1326–31.
Feldman M, Asselin M-C, Liu J, Wang S, McMahon A, Anton-Rodriguez J, Walker M, Symms M, Brown G, Hinz R, Matthews J, Bauer M, Langer O, Thom M, Jones T, Vollmar C, Duncan JS, Sisodiya SM, Koepp MJ. P-glycoprotein expression and function in patients with temporal lobe epilepsy: a case-control study. Lancet Neurol. 2013;12:777–85.
Volk HA, Löscher W. Multidrug resistance in epilepsy: rats with drug-resistant seizures exhibit enhanced brain expression of P-glycoprotein compared with rats with drug-responsive seizures. Brain. 2005;128:1358–68.
O’Brien FE, Clarke G, Fitzgerald P, Dinan TG, Griffin BT, Cryan JF. Inhibition of P-glycoprotein enhances transport of imipramine across the blood-brain barrier: microdialysis studies in conscious freely moving rats. Br J Pharmacol. 2012;166:1333–43.
de Klerk OL, Willemsen AT, Roosink M, Bartels AL, Hendrikse NH, Bosker FJ, den Boer JA. Locally increased P-glycoprotein function in major depression: a PET study with [11C]verapamil as a probe for P-glycoprotein function in the blood-brain barrier. Int J Neuropsychopharmacol. 2009;12:895–904.
Kim RB, Fromm MF, Wandel C, Leake B, Wood AJ, Roden DM, Wilkinson GR. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-protease inhibitors. J Clin Invest. 1998;101:289–94.
Robillard KR, Chan GNY, Zhang G, la Porte C, Cameron W, Bendayan R. Role of P-glycoprotein in the distribution of the HIV protease inhibitor atazanavir in the brain and male genital tract. Antimicrob Agents Chemother. 2014;58:1713–22.
Shaik N, Giri N, Pan G, Elmquist WF. P-glycoprotein-mediated active efflux of the anti-HIV1 nucleoside abacavir limits cellular accumulation and brain distribution. Drug Metab Dispos. 2007;35:2076–85.
Chai AB, Leung GKF, Callaghan R, Gelissen IC. P-glycoprotein: a role in the export of amyloid-β in Alzheimer's disease? FEBS J. 2020;287:612–25.
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman JR. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science. 2010;330:1774.
Vogelgesang S, Cascorbi I, Schroeder E, Pahnke J, Kroemer HK, Siegmund W, Kunert-Keil C, Walker LC, Warzok RW. Deposition of Alzheimer’s β-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics. 2002;12:533–41.
Wijesuriya HC, Bullock JY, Faull RLM, Hladky SB, Barrand MA. ABC efflux transporters in brain vasculature of Alzheimer’s subjects. Brain Res. 2010;1358:228–38.
Deo AK, Borson S, Link JM, Domino K, Eary JF, Ke B, Richards TL, Mankoff DA, Minoshima S, O’Sullivan F, Eyal S, Hsiao P, Maravilla K, Unadkat JD. Activity of p-glycoprotein, a β-amyloid transporter at the blood-brain barrier, is compromised in patients with mild Alzheimer disease. J Nucl Med. 2014;55:1106–11.
van Assema DME, Lubberink M, Bauer M, van der Filer WM, Schuit RC, Windhorst AD, Comans EFI, Hoetjes NJ, Tolboom N, Langer O, Müller M, Scheltens P, Lammertsma AA, van Berckel BNM. Blood-brain barrier P-glycoprotein function in Alzheimer’s disease. Brain. 2012;135:181–9.
Zoufal V, Mairinger S, Brackhan M, Krohn M, Filip T, Sauberer M, Stanek J, Wanek T, Tournier N, Bauer M, Pahnke J, Langer O. Imaging P-glycoprotein induction at the blood-brain barrier of a beta-amyloidosis mouse model with 11C-metoclopramide PET. J Nucl Med. 2019 Dec 5. jnumed.119.237198. doi: 10.2967/jnumed.119.237198.
Bartels AL, Willemsen ATM, Kortekaas R, de Jong BM, de Vries R, de Klerk O, van Oostrom JCH, Portman A, Leenders KL. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J Neural Transm (Vienna). 2008;115:1001–1009.
Vautier S, Fernandez C. ABCB1: the role in Parkinson’s disease and pharmacokinetics of antiparkinsonian drugs. Expert Opin Drug Metab Toxicol. 2009;5:1349–58.
Li Y, Li Y, Pang S, Huang W, Zhang A, Hawley RG, Yan B. Novel and functional ABCB1 gene variant in sporadic Parkinson’s disease. Neurosci Lett. 2014;566:61–6.
Luurtsema G, Elsinga P, Dierckx R, Boellaard R, van Waarde A. PET tracers for imaging of ABC transporters at the blood-brain barrier: principles and strategies. Curr Pharm Des. 2016;22:5779–85.
Raaphorst RM, Windhorst AD, Elsinga PH, Colabufo NA, Lammertsma AA, Luurtsema G. Radiopharmaceuticals for assessing ABC transporters at the blood-brain barrier. Clin Pharmacol Ther. 2015;97:362–71.
Lubberink M. Kinetic models for measuring P-glycoprotein function at the blood-brain barrier with positron emission tomography. Curr Pharm Des. 2016;22:5786–92.
Kannan P, John C, Zoghbi SS, Halldin C, Gottesman MM, Innis RB, Hall MD. Imaging the function of P-glycoprotein with radiotracers: pharmacokinetics and in vivo applications. Clin Pharmacol Thr. 2009;86:368–77.
Luurtsema G, Molthoff CFM, Schuit RC, Windhorst AD, Lammertsma AA, Franssen EJF. Evaluation of (R)-[11C]verapamil as PET tracer of P-glycoprotein function in the blood-brain barrier: kinetics and metabolism in the rat. Nucl Med Biol. 2005;32:87–93.
Savolainen H, Cantore M, Colabufo NA, Elsinga PH, Windhorst AD, Luurtesma G. Synthesis and preclinical evaluation of three novel fluorine-18 labeled radiopharmaceuticals for P-glycoprotein PET imaging at the blood-brain barrier. Mol Pharm. 2015;12:2265–75.
Savolainen H, Windhorst AD, Elsinga PH, Cantore M, Colabufo NA, Willemsen AT, Luurtsema G. Evaluation of [18F]MC225 as a PET radiotracer for measuring P-glycoprotein function at the blood-brain barrier in rats: kinetics, metabolism, and selectivity. J Cereb Blood Flow Metab. 2017;37:1286–98.
Savolainen H, Meerlo P, Elsinga PH, Windhorst AD, Dierckx RAJO, Colabufo NA, van Waarde A, Luurtsema G. P-glycoprotein function in the rodent brain displays a daily rhythm, a quantitative in vivo PET study. AAPS J. 2016;18:1524–31.
Toyohara J, García-Varela L, Kakiuchi T, Ohba H, Nishiyama S, Tago T, Vállez-García D, Boellaard R, Elsinga PH, Tsukada H, Luurtsema G. Head to head comparison of (R)-[11C]verapamil and [18F]MC225 in non-human primates; tracers for measuring P-gp function at the blood-brain barrier. Eur J Nucl Med Mol Imaging. 2017;44:S366 (abstract).
Toyohara J, Sakata M, Tago T, Colabufo N, Luurtsema G. Dosimetry and toxicology of [18F]MC225 for measuring P-glycoprotein function at the blood-brain barrier in humans. J Labelled Comp Radiopharm. 2019;62:S388–S389 (abstract).
Kirschner AS, Ice RD, Beierwaltes WH. Radiation dosimetry of 131I-19-iodocholesterol: the pitfalls of using tissue concentration data–reply. J Nucl Med. 1975;16:248–9.
Stabin MG, Sparks RB, Crowe E. OLINDA/EXM: the second-generation personal computer software for internal dose assessment in nuclear medicine. J Nucl Med. 2005;46:1023–7.
ICRP, 2007. The 2007 recommendations of the international commission on radiological protection. ICRP publication 103. Ann ICRP. 2007;37:1–332.
Fusi F, Durante M, Gorelli B, Perrone MG, Colabufo NA, Saponara S. MC225, a novel probe for P-glycoprotein PET imaging at the blood-brain barrier: in vitro cardiovascular safety evaluation. J Cardiovasc Pharmacol. 2017;70:405–10.
Koziorowski J, Behe M, Decristoforo C, Ballinger C, Elsinga P, Ferrari V, Kolenc Peitl P, Todde S, Mindt TL. Position paper on requirements for toxicological studies in the specific case of radiopharmaceuticals. EJNMMI Radiopharm Chem. 2016;1:1.
Zanotti-Fregonara P, Lammertsma AA, Innis RB. Suggested pathway to assess radiation safety of 18F-labeled PET tracers for first-in-human studies. Eur J Nucl Med Mol Imaging. 2013;40:1781–3.
ICRP, 1992. Radiological protection in biomedical research. ICRP publication 62. Ann ICRP. 1992;22:1–18.
RDRC Final Guidance: Human research without an investigational new drug application (August 2010). Available at: https://www.fda.gov/media/76286/download.
Sattler B, Kranz M, Starke A, Wilke S, Donat CK, Deuther-Conard W, Patt M, Schildan A, Patt J, Smits R, Hoepping A, Schoenknecht P, Steinbach J, Brust P, Sabri O. Internal dose assessment of (–)-18F-flubatine, comparing animal model datasets of mice and piglets with first-in-human results. J Nucl Med. 2014;55:1885–92.
Acknowledgements
We with the thank Mr. Kunpei Hayashi and Mr. Masanari Sakai for their technical support with cyclotron operation and radiosynthesis.
Funding
This work was supported in part by a Grant-in-Aid for Scientific Research (C) No. 18K07658 from the Japan Society for the Promotion of Science, and an Internal Research Grant for Translational Research from the Tokyo Metropolitan Institute of Gerontology (TR29-01).
Author information
Authors and Affiliations
Contributions
JT and GL conceived and designed the experiments. JT and TT performed the experiments. JT and MS analyzed the data. NC and GL contributed the reagents/materials/analysis tools. JT, NC, and GL wrote the paper. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All applicable institutional and/or national guidelines for the care and use of animals were followed, and the study protocol was approved by the institutional Research Ethics Board.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Toyohara, J., Sakata, M., Tago, T. et al. Automated synthesis, preclinical toxicity, and radiation dosimetry of [18F]MC225 for clinical use: a tracer for measuring P-glycoprotein function at the blood-brain barrier. EJNMMI Res 10, 84 (2020). https://doi.org/10.1186/s13550-020-00674-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13550-020-00674-6