
Abstract
Background
Loss of sarcolemmal nNOSμ is a common manifestation in a wide variety of muscle diseases and contributes to the dysregulation of multiple muscle activities. Given the critical role sarcolemmal nNOSμ plays in muscle, restoration of sarcolemmal nNOSμ should be considered as an important therapeutic goal.
Methods
nNOSμ is anchored to the sarcolemma by dystrophin spectrin-like repeats 16 and 17 (R16/17) and the syntrophin PDZ domain (Syn PDZ). To develop a strategy that can independently restore sarcolemmal nNOSμ, we engineered an R16/17-Syn PDZ fusion construct and tested whether this construct alone is sufficient to anchor nNOSμ to the sarcolemma in three different mouse models of Duchenne muscular dystrophy (DMD).
Results
Membrane-associated nNOSμ is completely lost in DMD. Adeno-associated virus (AAV)-mediated delivery of the R16/17-Syn PDZ fusion construct successfully restored sarcolemmal nNOSμ in all three models. Further, nNOS restoration was independent of the dystrophin-associated protein complex.
Conclusions
Our results suggest that the R16/17-Syn PDZ fusion construct is sufficient to restore sarcolemmal nNOSμ in the dystrophin-null muscle.
Similar content being viewed by others


Background
Nitric oxide synthases (NOS) catalyze the production of the signaling messenger, nitric oxide (NO). Neuronal NOS (nNOS) is the primary NOS isoform in skeletal muscle [1, 2]. nNOSμ is the primary nNOS isoform in muscle and it is localized at the sarcolemma. Sarcolemmal nNOSμ plays an important role in regulating multifaceted activities of muscle, including blood perfusion [3, 4], glucose metabolism [5,6,7,8], oxidative stress [9, 10], muscle contractility [11, 12], muscle satellite cell activation and muscle repair [13,14,15,16,17,18], mitochondria biogenesis [19,20,21,22], muscle mass [23,24,25,26], and muscle fatigue [26,27,28,29].
Activation of nNOSμ is dependent on dimerization of nNOSμ proteins. Each monomer contains a PDZ domain, an oxygenase domain, a calmodulin-binding site, and a reductase domain [30]. The interactions of two oxygenase domains mediate nNOSμ dimer formation and enzymatic activation [31, 32]. Loss of sarcolemmal nNOSμ impairs multiple NO-mediated activities [3, 4, 23, 29, 33]. nNOSμ localization at the sarcolemma depends on dystrophin and α1-syntrophin [34, 35]. Previously, biochemistry and X-ray crystallography studies have revealed that the α1-syntrophin PDZ domain binds to the nNOSμ PDZ domain [36, 37]. Transgenic studies further confirmed that the interaction between α1-syntrophin and nNOSμ is crucial for nNOSμ localization at the sarcolemma [3, 34, 38]. However, α1-syntrophin alone is not sufficient to anchor nNOSμ to the sarcolemma [35]. We have previously shown that dystrophin spectrin-like repeats 16 and 17 (R16/17) are essential for sarcolemmal localization of nNOSμ [39, 40]. Multiple lines of evidence, including yeast two-hybrid assay, biochemical and molecular analysis, and structural modeling, suggest that dystrophin R16/17 directly interact with nNOSμ PDZ domain [39,40,41,42,43]. Dystrophin is a sub-sarcolemmal protein. It maintains sarcolemmal integrity during muscle contraction and organizes a group of transmembrane and cytosolic proteins (such as dystroglycans, sarcoglycans, sarcospan, syntrophins, dystrobrevin, and nNOSμ) into the dystrophin-associated protein complex (DAPC) at the sarcolemma [44, 45].
Loss and/or diminished expression of dystrophin or DAPC components leads to a variety of muscular dystrophies such as Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD), and several forms of recessive limb-girdle muscular dystrophies (LGMD). Loss of sarcolemmal nNOS is a common feature in these muscular dystrophies [1, 35, 46,47,48,49]. Absence or reduction of sarcolemmal nNOS has also been reported in non-DAPC-related muscular dystrophies (eg. LGMD2B, MDC1A) [29, 50], inflammatory myopathies [50], cachexia [51], myasthenia gravis [26], diabetes [52], and aging-related muscle atrophy [53]. Collectively, accumulated evidence suggests that loss of sarcolemmal nNOSμ is a common manifestation in a wide variety of muscle diseases. Importantly, the absence of sarcolemmal nNOSμ has been shown to either directly or indirectly contribute to the initiation and progression of these diseases. Given the critical role sarcolemmal nNOSμ plays in various muscle activities, restoration of sarcolemmal nNOSμ should be considered as an important therapeutic goal.
Since dystrophin R16/17 and the syntrophin PDZ domain are both required for nNOSμ localization at the sarcolemma [36, 40], we hypothesize that a fusion protein consisting of dystrophin R16/17 and the syntrophin PDZ domain can anchor nNOSμ to the sarcolemma. In this study, we tested our hypothesis by expressing a membrane-bound dystrophin R16/17-syntrophin PDZ fusion protein with adeno-associated virus (AAV) in dystrophin-null muscle. This approach successfully restored sarcolemmal nNOSμ.
Methods
Animal care and studies
All animal procedures were carried out in accordance with NIH guidelines, and all animal experiments were approved by the Animal Care and Use Committee (ACUC) of the University of Missouri. BL10 mice (stock #: 000666), Mdx (stock #: 001801), DBA/2 J-mdx (stock #: 013141), and heterozygous Cmah/mdx (stock #: 017929) mice were purchased from The Jackson Laboratory. Homozygous Cmah/mdx mice were generated by breeding heterozygous Cmah/mdx mice. Both male and female mice were used in this study. All the mice are maintained in a specific-pathogen-free animal care facility with access to food and water ad libitum.
Construct design
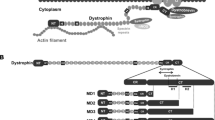
In a previous study, we engineered an AAV construct (YL299) that carries the expression cassette of dystrophin R16/17.GFP with a membrane-targeting motif (Pal) [40]. The Pal motif is derived from the Ras palmitoylation sequence and has been successfully used to target nNOSμ, α-dystrobrevin-2a, and dystrophin R16/17 to the muscle membrane [40, 54, 55]. Here, we used YL299 as the backbone and inserted syntrophin PDZ domain between R17 and GFP. The linker sequence GGSG was included to connect dystrophin R16/17 and the syntrophin PDZ domain (Fig. 1a and Additional file 1: Figure S1). The syntrophin PDZ sequence was engineered into pYL299 by PCR-based cloning method using the full-length mouse syntrophin cDNA plasmid as the template (a gift from Dr. Stanley C. Froehner, University of Washington, Seattle, WA, USA). The resulting construct was named as YL465. In YL465, the expression of R16/17.Syn PDZ.GFP.Pal (Fig. 1a) was regulated by the CMV promoter and SV40 polyadenylation signal.

Dystrophin R16/17-syntrophin PDZ domain fusion protein restored sarcolemmal nNOSμ in mdx mice. a. Schematic outline of the fusion construct. R16/17, dystrophin spectrin-like repeats 16 and 17; Syn PDZ, syntrophin PDZ domain; Pal, the palmitoylation motif for membrane targeting. b. AAV viruses were injected into both TA muscles of six mdx mice (n = 6). Representative photomicrographs of serial muscle sections visualized for GFP, R17, syntrophin PDZ domain, and nNOS expression and nNOS activity from wild-type BL10 mice, untreated mdx mice, and AAV.R16/17-Syn PDZ.GFP.Pal treated mdx mice. Asterisk, the same myofiber in serial sections. Scale bar = 50 μm
AAV production and injection
Recombinant AAV-9 viruses were produced using our published protocol, which involves triple-plasmid transfection in the human embryonic kidney (HEK) 293 cells and three rounds of CsCl ultracentrifugation purification [40, 56]. AAV titer was determined by real-time PCR using Fast SYBR Green Master Mix kit (Applied Biosystems, Foster City, CA) with a pair of primers that amplify a fragment in the CMV promoter: forward primer: 5′-TTACGGTAAACTGCCCACTTG-3′; reverse primer: 5′-CATAAGGTCATGTACTGGGCATAA-3′.
A total of 5 × 1011 viral genome (vg) particles of AAV vectors (in a volume of 30 μl) were injected into the tibialis anterior (TA) muscle of six adult (2 to 4-month-old) mdx, three DBA/2 J-mdx [57,58,59], and three Cmah/mdx mice [60] according to our established method [40]. Four weeks after AAV injection, TA muscles were harvested, embedded in Tissue-Tek OCT (Sakura Finetek), and snap-frozen in 2-methylbutane with liquid nitrogen.
Histology studies
Histology studies were performed on 10-μm cryosections of the TA muscles. General morphology of the muscle was examined by H.E. staining. GFP signal was detected by direct visualization under a fluorescence microscope. Dystrophin R17 domain was revealed by immunofluorescence staining with a mouse anti-R17 antibody (1:500, a gift from Dr. Glenn Morris, The Rober Jones and Agnes Hunt Orthopedic Hospital, Oswestry, Shropshire, United Kingdom). Sarcolemmal nNOSμ was identified by immunostaining with a rabbit anti-C-terminus of nNOS antibody (1:8000, N7280, Sigma), and nNOS activity staining was performed as we published before [56]. For nNOS activity staining, 16-μm cryosections were first fixed in 4% paraformaldehyde for 2 h at 4 °C. After a brief rinse in phosphate buffered saline (PBS), the tissue sections were permeabilized with 0.2% Triton X-100 at 37 °C for 20 min. The nicotinamide adenine dinucleotide phosphate (NADPH) diaphorase activity of nNOS was revealed by adding the mixture of 0.2% Triton X-100, 0.2 mM NADPH, and 0.16 mg/ml nitroblue tetrazolium (N6876-100MG, Sigma-Aldrich) [39, 40, 61]. nNOS activity appears as blue staining under the bright field. The syntrophin PDZ domain was detected by a mouse anti-pan-syntrophin antibody (1:500, ab11425, Abcam). β-dystroglycan was detected with a mouse anti-β-dystroglycan antibody (1:50, NCL-b-DG, Novocastra/Leica Biosystems). β-sarcoglycan was detected with a mouse anti-β-sarcoglycan antibody (1:50, NCL-b-SARC, Novocastra/Leica Biosystems). Dystrobrevin was detected with a mouse anti-dystrobrevin antibody (1:200, 610,766, BD Biosciences). At least three non-contiguous sections of each sample were examined by histology studies and representative images were present in the figures. In immunostaining, secondary antibody only was used as the negative control. Muscle histology was evaluated by two independent researchers, who were blinded for the information of experimental groups.
Western blot
The whole TA muscle was homogenized by mechanical disruption with a mortar and a pestle in liquid nitrogen. Then, muscle tissues were lysed in the lysis buffer containing 10% sodium dodecyl sulfate (SDS), 5 mM ethylenediaminetetraacetic acid (EDTA), 62.5 mM Tris.HCl (pH 6.8), plus 1% cocktail proteinase inhibitor (11836153001, Roche Applied Science). The whole muscle lysate was obtained after spinning at 14,000 rpm for 2 min. The membrane-enriched microsomal fraction was extracted with the Plasma Membrane Protein Extraction kit (ab65400, Abcam) according to the manufacturer instructions [62]. The muscle lysates were separated on 6% or 8% SDS-polyacrylamide gel and transferred to the polyvinylidene difluoride (PVDF) membrane. The PVDF membranes were probed with the following antibodies: mouse anti-R17 antibody (1:500, a gift from Dr. Glenn Morris), mouse anti-pan-syntrophin antibody (1:600, ab11425, Abcam), rabbit anti-C-terminus of nNOS antibody (1:2000, N7280, Sigma), and rabbit anti-GFP antibody (1:1000, ab32146, Abcam). The protein loading was confirmed with an antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:5000, MAB374, Millipore) for the whole lysate and an antibody against α1-Na+/K+ATPase (1:3000, ab7671, Abcam) for the microsomal fraction.
Quantification of immunoblotting was achieved by ImageStudioLite (Li-Cor Biosciences) according to the manufacturer’s instructions. Three different blots were quantified, and the signal of nNOS is normalized to that of GAPDH. The data were analyzed with the program GraphPad Prism 6.0a for Mac OS X (GraphPad Software, La Jolla, CA, USA). Comparison among three groups was done by ANOVA. Tukey’s test was used as the post hoc test to compare the difference between the two groups. The statistical significance was considered when the P value is less than 0.05.
Results
Membrane-bound R16/17-Syn PDZ fusion protein restored sarcolemmal nNOSμ in mdx muscle
We expressed the R16/17-Syn PDZ fusion protein (Fig. 1a) with AAV in mdx muscle. Expression of the fusion protein was characterized by the GFP signal and immunostaining with antibodies against R17 and the syntrophin PDZ domain. As shown in Fig. 1b, AAV delivery resulted in the efficient expression of the fusion protein at the sarcolemma in the dystrophic muscle. As revealed by positive nNOS immunostaining and activity staining at the sarcolemma, expression of the membrane-bound fusion protein successfully restored sarcolemmal nNOSμ in the dystrophin-null muscle (Fig. 1b).
Western blot was performed to corroborate the expression of the fusion protein and restoration of sarcolemmal nNOSμ. The robust expression of the fusion protein was confirmed by immunoblotting with antibodies against R17, GFP, and the syntrophin PDZ domain in the whole muscle lysate (Fig. 2a). Consistent with previous studies [1, 12], dystrophin deficiency significantly reduced the total amount of nNOSμ in the muscle. AAV-mediated expression of the R16/17-Syn PDZ did not change the total nNOSμ level in mdx muscle (Fig. 2a). Membrane localization of the fusion protein was validated by microsomal preparation western blot with antibodies against R17, GFP, and the syntrophin PDZ domain (Fig. 2b). Importantly, restoration of sarcolemmal nNOSμ was confirmed by identifying nNOSμ in the microsomal fraction of the AAV-injected mdx muscle (Fig. 2b).

Immunoblot investigation of R16/17-Syn PDZ fusion protein expression and restoration of sarcolemmal nNOSμ in mdx mice. a. Whole muscle lysate western blot. The robust expression of the fusion protein was revealed by antibodies against R17, GFP, and the syntrophin PDZ domain. The total amount of nNOS in the AAV-injected muscle is almost the same as that of uninjected mdx muscle. Quantification of band intensity confirmed that the total amount of nNOS in the wild-type muscle is significantly higher than in mdx and AAV-injected muscles (asterisk, significantly different from mdx or AAV-injected muscle). GAPDH is the loading control. b. Microsomal preparation western blot. The membrane expression of the fusion protein was detected by antibodies against R17, GFP, and the syntrophin PDZ domain. Restoration of sarcolemmal nNOSμ by the fusion protein was confirmed by identifying nNOS in the microsomal preparation of AAV-injected muscle. ATPase is the loading control. Open arrowhead, full-length dystrophin; filled arrowhead, R16/17-Syn PDZ fusion protein; arrow, endogenous syntrophin
Restoration of sarcolemmal nNOSμ by the fusion protein was independent of the DAPC
Next, we analyzed whether restoration of sarcolemmal nNOSμ by the R16/17-Syn PDZ fusion protein requires the presence of other DAPC components. As reported previously [63, 64], sarcolemmal expression of β-dystroglycan, β-sarcoglycan, dystrobrevin, syntrophins, and nNOS was greatly reduced in uninjected mdx muscle (Fig. 3). In the serial sections of AAV-injected muscle, GFP signal, syntrophin staining, and nNOS staining showed sarcolemmal expression of the fusion protein and restoration of nNOSμ sarcolemmal localization. However, immunostaining for β-dystroglycan, β-sarcoglycan, and dystrobrevin showed similar levels of expression as that of uninjected mdx muscle (Fig. 3).

Restoration of sarcolemmal nNOSμ by the R16/17-Syn PDZ fusion protein was independent of the DAPC. Representative photomicrographs of serial muscle sections visualized for GFP, syntrophin PDZ domain, nNOS expression, β-dystroglycan (β-DG), β-sarcoglycan (β-SG), and dystrobrevin (DB) in uninjected mdx mice and AAV.R16/17-Syn PDZ.GFP.Pal treated mdx mice. Asterisk, the same myofiber in serial sections. Scale bar = 50 μm
The fusion protein also restored sarcolemmal nNOSμ in Cmah/mdx and DBA/2 J-mdx mice
Cmah/mdx and DBA/2 J-mdx mice are two severe mouse DMD models [57,58,59,60]. In addition to the mutation in the dystrophin gene, Cmah/mdx mice also carry an inactivating deletion in the CMAH (cytidine monophosphate sialic acid hydroxylase) gene [60]. DBA/2 J-mdx congenic mice were generated by backcrossing C57BL/10-mdx with DBA-2 J inbred mice for several generations. To test whether our fusion construct can restore sarcolemmal nNOSμ localization in mice with more severe phenotypes, we performed AAV injection in Cmah/mdx and DBA/2 J-mdx mice. Similar to that of mdx mice (Figs. 1 and 3), AAV injection resulted in the expression of the R16/17-Syn PDZ fusion protein at the sarcolemma (Fig. 4). nNOS immunostaining and activity staining showed the recovery of sarcolemmal nNOSμ in serial muscle sections (Fig. 4).

The R16/17-Syn PDZ fusion protein restored sarcolemmal nNOSμ in Cmah/mdx and DBA/2 J-mdx mice. AAV viruses were injected into both TA muscles of three Cmah/mdx (n = 3) and three DBA/2 J-mdx mice (n = 3). Representative photomicrographs of serial muscle sections visualized for GFP, R17, syntrophin (Syn), nNOS expression, and nNOS activity staining. In non-transduced myofibers, there is no sarcolemmal nNOSμ restoration. Asterisk, the same myofiber in serial sections; square, the non-transduced myofiber. Scale bar = 50 μm
Discussion
In this study, we engineered a fusion protein consisting of the minimal components required for sarcolemmal nNOSμ localization and expressed the fusion protein with AAV gene transfer in dystrophin-null mice. Microscopic examination revealed successful expression of the fusion protein at the sarcolemma and restoration of membrane-bound nNOSμ (Figs. 1, 2, and 4). These findings were confirmed by whole muscle lysate and microsomal preparation western blot (Figs. 1 and 2). We also showed that restoration of sarcolemmal nNOSμ by the fusion protein was independent of other components of the DAPC (Fig. 3).
In our previous studies, we demonstrated that dystrophin R16/17 serves as the nNOS-binding domain in the context of a dystrophin protein [39, 40]. We tested a series of R16/17-containing mini- and micro-dystrophins. These synthetic R16/17-inclusive dystrophins successfully restore sarcolemmal nNOSμ expression in the murine and canine DMD models and offer better protection than those without R16/17 in animal models [39, 65,66,67,68,69,70]. An R16/17-inclusive micro-dystrophin has already been used in a clinical trial [71]. However, it remained elusive whether R16/17 alone can restore sarcolemmal nNOSμ in the dystrophin-null muscle. We have previously tested AAV constructs that expressed dystrophin R16/17 with and without Pal motif. Without Pal motif, R16/17 are confined to the cytosol of myofibers, while with Pal motif, R16/17 are targeted to the muscle membrane [40]. In mdx mice, membrane-bound R16/17 did not restore sarcolemmal nNOSμ. However, the exact same construct successfully restored sarcolemmal nNOSμ in ΔH2-R19 mini-dystrophin transgenic mice [40]. R16/17 are missing in both mdx mice and ΔH2-R19 mini-dystrophin transgenic mice. Most sarcolemmal DAPC components (dystroglycans, sarcoglycans, syntrophin, and dystrobrevin but not nNOS) were restored in transgenic mice but not mdx mice [63, 64, 72, 73]. Since syntrophin has been shown to interact with nNOSμ [36, 37], we reasoned that successful restoration of sarcolemmal nNOSμ in transgenic mice was likely due to the presence of syntrophin at the sarcolemma in transgenic mice. Similarly, we speculated that failure to restore sarcolemmal nNOSμ in mdx mice by membrane-bound R16/17 was due to the lack of sarcolemmal syntrophin [63, 64, 72, 73]. However, several important issues remain unclear. First, it is not clear whether other DAPC components (dystroglycans, sarcoglycans, and dystrobrevin) have contributed to the successful restoration of sarcolemmal nNOSμ in transgenic mice. Second, since dystrophic phenotype was largely attenuated in transgenic mice, it is unclear whether the improved muscle microenvironment also plays a role. To address these questions, we now generated the membrane-bound R16/17-Syn PDZ construct and tested it in mildly affected mdx mice and in Cmah/mdx and DBA/2 J-mdx mice, two severely affected DMD mouse models. Although the membrane-bound R16/17-Syn PDZ construct did not restore other DAPC components (such as β-dystroglycan, β-sarcoglycan, and dystrobrevin) (Fig. 3), nor was the muscle disease attenuated, the fusion construct successfully restored sarcolemmal nNOSμ. These results suggest that R16/17 and the syntrophin PDZ domain are the only components required for the sarcolemmal localization of nNOSμ. Other DAPC components and muscle microenvironment have a nominal impact.
In all the published studies, R16/17 and syntrophin are provided separately as two independent molecules. From a drug development standpoint, this is less appealing. Ideally, one would want to put both R16/17 and the syntrophin PDZ domain together as one molecule. However, putting R16/17 next to the syntrophin PDZ domain in a single molecule may create a spatial hindrance for the simultaneous interaction of nNOSμ with both R16/17 and the syntrophin PDZ domain. Our results suggest that this is not an issue (Fig. 5). Dimerization of nNOSμ is essential for its activity. Although both R16/17 and syntrophin PDZ directly interact with nNOSμ PDZ domain, it remains to be determined whether the single PDZ domain of a nNOSμ monomer bind to both dystrophin R16/17 and syntrophin PDZ domain, or two different PDZ domains of an activated nNOS dimer interact with R16/17 and syntrophin PDZ separately, namely one nNOSμ PDZ domain interacts with R16/17 and the other nNOSμ PDZ domain interacts with syntrophin (Fig. 5).

The model of sarcolemmal nNOS restoration by R16/17-Syn PDZ fusion protein. The R16/17-Syn PDZ fusion protein is targeted to the sarcolemma by the membrane-targeting motif (Pal). R16/17 and the syntrophin PDZ domain in the fusion protein interact with nNOSμ PDZ domain to anchor nNOSμ to the sarcolemma (not drawn to scale). Please note an activated nNOSμ exists as a dimer. It is currently unclear how exactly the two PDZ domains in the nNOSμ dimer interact with dystrophin R16/17 and the syntrophin PDZ domain. It is possible that one nNOSμ PDZ domain interacts with dystrophin R16/17 and the other nNOSμ PDZ domain interacts with the syntrophin PDZ domain. It is also possible that a single nNOSμ PDZ domain can interact with both dystrophin R16/17 and the syntrophin PDZ domain. For simplicity, the activated nNOSμ dimer was depicted as a single molecule in this cartoon
Our study has several limitations. First, we have only limited our investigation in mouse DMD models. Future studies in other disease models (such as cachexia and aging-related muscle atrophy, inflammatory myopathies, myasthenia gravis, and other muscular dystrophies that display sarcolemmal nNOSμ delocalization) will be helpful to determine whether our fusion construct can serve as a universal treatment to restore membrane-associated nNOSμ expression in these conditions. Second, our current study has mainly focused on the morphological and biochemical demonstration of sarcolemmal nNOSμ restoration. Future functional and physiological studies are needed to show improvements in nNOSμ regulated muscle activities and signaling pathways with the dystrophin R16/17-syntrophin PDZ fusion protein.
Conclusions
In this study, we found that the fusion protein containing dystrophin R16/17 and syntrophin PDZ domain restored sarcolemmal nNOSμ in dystrophin-null mice. Sarcolemmal nNOSμ restoration by the fusion protein is independent of the DAPC. This fusion construct has established the basis for developing a universal treatment to restore sarcolemmal nNOSμ in a wide variety of muscle diseases.
Abbreviations
- AAV:
-
Adeno-associated virus
- ACUC:
-
Animal care and use committee
- ANOVA:
-
Analysis of variance
- BMD:
-
Becker muscular dystrophy
- CMAH:
-
Cytidine monophosphate sialic acid hydroxylase
- CMV:
-
Cytomegalovirus
- DAPC:
-
Dystrophin-associated protein complex
- DMD:
-
Duchenne muscular dystrophy
- EDTA:
-
Ethylenediaminetetraacetic acid
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- HEK:
-
Human embryonic kidney
- LGMD:
-
Limb-girdle muscular dystrophy
- MDC1A:
-
Merosin-deficient congenital muscular dystrophy
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- nNOS:
-
Neuronal nitric oxide synthase
- NO:
-
Nitric oxide
- PBS:
-
Phosphate buffered saline
- PVDF:
-
Polyvinylidene difluoride
- SDS:
-
Sodium dodecyl sulfate
- TA:
-
Tibialis anterior
References
Brenman JE, Chao DS, Xia H, Aldape K, Bredt DS. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995;82:743–52.
Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81:209–37.
Thomas GD, Shaul PW, Yuhanna IS, Froehner SC, Adams ME. Vasomodulation by skeletal muscle-derived nitric oxide requires alpha-syntrophin-mediated sarcolemmal localization of neuronal nitric oxide synthase. Circ Res. 2003;92:554–60.
Thomas GD, Sander M, Lau KS, Huang PL, Stull JT, Victor RG. Impaired metabolic modulation of alpha-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc Natl Acad Sci U S A. 1998;95:15090–5.
Wehling-Henricks M, Oltmann M, Rinaldi C, Myung KH, Tidball JG. Loss of positive allosteric interactions between neuronal nitric oxide synthase and phosphofructokinase contributes to defects in glycolysis and increased fatigability in muscular dystrophy. Hum Mol Genet. 2009;18:3439–51.
Roberts CK, Barnard RJ, Scheck SH, Balon TW. Exercise-stimulated glucose transport in skeletal muscle is nitric oxide dependent. Am J Phys. 1997;273:E220–5.
Balon TW, Nadler JL. Evidence that nitric oxide increases glucose transport in skeletal muscle. J Appl Physiol. 1997;82:359–63.
Roy D, Perreault M, Marette A. Insulin stimulation of glucose uptake in skeletal muscles and adipose tissues in vivo is NO dependent. Am J Phys. 1998;274:E692–9.
Tews DS. Role of nitric oxide and nitric oxide synthases in experimental models of denervation and reinnervation. Microsc Res Tech. 2001;55:181–6.
Khan SA, Lee K, Minhas KM, Gonzalez DR, Raju SV, Tejani AD, et al. Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc Natl Acad Sci U S A. 2004;101:15944–8.
Percival JM, Anderson KN, Gregorevic P, Chamberlain JS, Froehner SC. Functional deficits in nNOSmu-deficient skeletal muscle: myopathy in nNOS knockout mice. PLoS One. 2008;3:e3387.
Li D, Yue Y, Lai Y, Hakim CH, Duan D. Nitrosative stress elicited by nNOSmicro delocalization inhibits muscle force in dystrophin-null mice. J Pathol. 2011;223:88–98.
Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol Rev. 2013;93:23–67.
Dumont NA, Bentzinger CF, Sincennes MC, Satellite Cells RMA. Skeletal muscle regeneration. Compr Physiol. 2015;5:1027–59.
Anderson JE. A role for nitric oxide in muscle repair: nitric oxide-mediated activation of muscle satellite cells. Mol Biol Cell. 2000;11:1859–74.
Dumont NA, Rudnicki MA. Targeting muscle stem cell intrinsic defects to treat Duchenne muscular dystrophy. NPJ Regen Med. 2016;1:16006.
Wozniak AC, Anderson JE. The dynamics of the nitric oxide release-transient from stretched muscle cells. Int J Biochem Cell Biol. 2009;41:625–31.
Buono R, Vantaggiato C, Pisa V, Azzoni E, Bassi MT, Brunelli S, et al. Nitric oxide sustains long term skeletal muscle regeneration by regulating satellite cells fate via signalling pathways requiring Vangl2 and cyclic GMP. Stem Cells. 2012;30:197–209.
Wadley GD, Choate J, McConell GK. NOS isoform-specific regulation of basal but not exercise-induced mitochondrial biogenesis in mouse skeletal muscle. J Physiol. 2007;585:253–62.
Schild L, Jaroscakova I, Lendeckel U, Wolf G, Keilhoff G. Neuronal nitric oxide synthase controls enzyme activity pattern of mitochondria and lipid metabolism. FASEB J. 2006;20:145–7.
Tengan CH, Rodrigues GS, Godinho RO. Nitric oxide in skeletal muscle: role on mitochondrial biogenesis and function. Int J Mol Sci. 2012;13:17160–84.
De Palma C, Morisi F, Pambianco S, Assi E, Touvier T, Russo S, et al. Deficient nitric oxide signalling impairs skeletal muscle growth and performance: involvement of mitochondrial dysregulation. Skelet Muscle. 2014;4:22.
Suzuki N, Motohashi N, Uezumi A, Fukada S, Yoshimura T, Itoyama Y, et al. NO production results in suspension-induced muscle atrophy through dislocation of neuronal NOS. J Clin Invest. 2007;117:2468–76.
Pietri-Rouxel F, Gentil C, Vassilopoulos S, Baas D, Mouisel E, Ferry A, et al. DHPR alpha1S subunit controls skeletal muscle mass and morphogenesis. EMBO J. 2010;29:643–54.
Lawler JM, Kunst M, Hord JM, Lee Y, Joshi K, Botchlett RE, et al. EUK-134 ameliorates nNOSmu translocation and skeletal muscle fiber atrophy during short-term mechanical unloading. Am J Physiol Regul Integr Comp Physiol. 2014;306:R470–82.
Meinen S, Lin S, Rüegg MA, Punga AR. Fatigue and muscle atrophy in a mouse model of myasthenia gravis is paralleled by loss of sarcolemmal nNOS. PLoS One. 2012;7:e44148.
Percival JM. nNOS regulation of skeletal muscle fatigue and exercise performance. Biophys Rev. 2011;3:209–17.
Percival JM, Anderson KN, Huang P, Adams ME, Froehner SC. Golgi and sarcolemmal neuronal NOS differentially regulate contraction-induced fatigue and vasoconstriction in exercising mouse skeletal muscle. J Clin Invest. 2010;120:816–26.
Kobayashi YM, Rader EP, Crawford RW, Iyengar NK, Thedens DR, Faulkner JA, et al. Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature. 2008;456:511–5.
Snyder SH, Ferris CD. Novel neurotransmitters and their neuropsychiatric relevance. Am J Psychiatry. 2000;157:1738–51.
Panda K, Rosenfeld RJ, Ghosh S, Meade AL, Getzoff ED, Stuehr DJ. Distinct dimer interaction and regulation in nitric-oxide synthase types I, II, and III. J Biol Chem. 2002;277:31020–30.
Zhou L, Zhu DY. Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide. 2009;20:223–30.
Sander M, Chavoshan B, Harris SA, Iannaccone ST, Stull JT, Thomas GD, et al. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 2000;97:13818–23.
Adams ME, Mueller HA, Froehner SC. In vivo requirement of the alpha-syntrophin PDZ domain for the sarcolemmal localization of nNOS and aquaporin-4. J Cell Biol. 2001;155:113–22.
Chao DS, Gorospe JR, Brenman JE, Rafael JA, Peters MF, Froehner SC, et al. Selective loss of sarcolemmal nitric oxide synthase in Becker muscular dystrophy. J Exp Med. 1996;184:609–18.
Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–67.
Hillier BJ, Christopherson KS, Prehoda KE, Bredt DS, Lim WA. Unexpected modes of PDZ domain scaffolding revealed by structure of nNOS-syntrophin complex. Science. 1999;284:812–5.
Kameya S, Miyagoe Y, Nonaka I, Ikemoto T, Endo M, Hanaoka K, et al. alpha1-syntrophin gene disruption results in the absence of neuronal-type nitric-oxide synthase at the sarcolemma but does not induce muscle degeneration. J Biol Chem. 1999;274:2193–200.
Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C, et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest. 2009;119:624–35.
Lai Y, Zhao J, Yue Y, Duan D. alpha2 and alpha3 helices of dystrophin R16 and R17 frame a microdomain in the alpha1 helix of dystrophin R17 for neuronal NOS binding. Proc Natl Acad Sci U S A. 2013;110:525–30.
Delalande O, Molza AE, Dos Santos-Morais R, Chéron A, Pollet É, Raguenes-Nicol C, et al. Dystrophin’s central domain forms a complex filament that becomes disorganized by in-frame deletions. J Biol Chem. 2018;293:6637–46.
Giudice E, Molza AE, Laurin Y, Nicolas A, Le Rumeur E, Delalande O. Molecular clues to the dystrophin-nNOS interaction: a theoretical approach. Biochemistry. 2013;52:7777–84.
Molza AE, Mangat K, Le Rumeur E, Hubert JF, Menhart N, Delalande O. Structural basis of neuronal nitric-oxide synthase interaction with dystrophin repeats 16 and 17. J Biol Chem. 2015;290:29531–41.
Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol. 2006;7:762–73.
Allen DG, Whitehead NP, Froehner SC. Absence of dystrophin disrupts skeletal muscle signaling: roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol Rev. 2016;96:253–305.
Torelli S, Brown SC, Jimenez-Mallebrera C, Feng L, Muntoni F, Sewry CA. Absence of neuronal nitric oxide synthase (nNOS) as a pathological marker for the diagnosis of Becker muscular dystrophy with rod domain deletions. Neuropathol Appl Neurobiol. 2004;30:540–5.
Gentil C, Leturcq F, Ben Yaou R, Kaplan JC, Laforet P, Penisson-Besnier I, et al. Variable phenotype of del45-55 Becker patients correlated with nNOSmu mislocalization and RYR1 hypernitrosylation. Hum Mol Genet. 2012;21:3449–60.
Crosbie RH, Barresi R, Campbell KP. Loss of sarcolemma nNOS in sarcoglycan-deficient muscle. FASEB J. 2002;16:1786–91.
Grady RM, Grange RW, Lau KS, Maimone MM, Nichol MC, Stull JT, et al. Role for alpha-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat Cell Biol. 1999;1:215–20.
Finanger Hedderick EL, Simmers JL, Soleimani A, Andres-Mateos E, Marx R, Files DC, et al. Loss of sarcolemmal nNOS is common in acquired and inherited neuromuscular disorders. Neurology. 2011;76:960–7.
Acharyya S, Butchbach ME, Sahenk Z, Wang H, Saji M, Carathers M, et al. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell. 2005;8:421–32.
Mezghenna K, Leroy J, Azay-Milhau J, Tousch D, Castex F, Gervais S, et al. Counteracting neuronal nitric oxide synthase proteasomal degradation improves glucose transport in insulin-resistant skeletal muscle from Zucker fa/fa rats. Diabetologia. 2014;57:177–86.
Samengo G, Avik A, Fedor B, Whittaker D, Myung KH, Wehling-Henricks M, et al. Age-related loss of nitric oxide synthase in skeletal muscle causes reductions in calpain S-nitrosylation that increase myofibril degradation and sarcopenia. Aging Cell. 2012;11:1036–45.
Rebolledo DL, Kim MJ, Whitehead NP, Adams ME, Froehner SC. Sarcolemmal targeting of nNOSμ improves contractile function of mdx muscle. Hum Mol Genet. 2016;25:158–66.
Adams ME, Tesch Y, Percival JM, Albrecht DE, Conhaim JI, Anderson K, et al. Differential targeting of nNOS and AQP4 to dystrophin-deficient sarcolemma by membrane-directed {alpha}-dystrobrevin. J Cell Sci. 2008;121:48–54.
Lai Y, Zhao J, Yue Y, Wasala NB, Duan D. Partial restoration of cardiac function with ΔPDZ nNOS in aged mdx model of Duchenne cardiomyopathy. Hum Mol Genet. 2014;23:3189–99.
Rodrigues M, Echigoya Y, Maruyama R, Lim KR, Fukada SI, Yokota T. Impaired regenerative capacity and lower revertant fibre expansion in dystrophin-deficient mdx muscles on DBA/2 background. Sci Rep. 2016;6:38371.
Coley WD, Bogdanik L, Vila MC, Yu Q, van der Meulen JH, Rayavarapu S, et al. Effect of genetic background on the dystrophic phenotype in mdx mice. Hum Mol Genet. 2016;25:130–45.
Fukada SI, Morikawa D, Yamamoto Y, Yoshida T, Sumie N, Yamaguchi M, et al. Genetic background affects properties of satellite cells and mdx phenotypes. Am J Pathol. 2010;176:2414–24.
Chandrasekharan K, Yoon JH, Xu Y, deVries S, Camboni M, Janssen PM, et al. A human-specific deletion in mouse Cmah increases disease severity in the mdx model of Duchenne muscular dystrophy. Sci Transl Med. 2010;2:42ra54.
Hope BT, Michael GJ, Knigge KM, Vincent SR. Neuronal NADPH diaphorase is a nitric oxide synthase. Proc Natl Acad Sci U S A. 1991;88:2811–4.
Zhao J, Kodippili K, Yue Y, Hakim CH, Wasala L, Pan X, et al. Dystrophin contains multiple independent membrane-binding domains. Hum Mol Genet. 2016;25:3647–53.
Judge LM, Haraguchiln M, Chamberlain JS. Dissecting the signaling and mechanical functions of the dystrophin-glycoprotein complex. J Cell Sci. 2006;119:1537–46.
Yue Y, Liu M, Duan D. C-terminal-truncated microdystrophin recruits dystrobrevin and syntrophin to the dystrophin-associated glycoprotein complex and reduces muscular dystrophy in symptomatic utrophin/dystrophin double-knockout mice. Mol Ther. 2006;14:79–87.
Zhang Y, Dongsheng D. Novel mini-dystrophin gene dual AAV vectors restore nNOS expression at the sarcolemma. Hum Gene Ther. 2012;23:98–103.
Hakim CH, Wasala NB, Pan X, Kodippili K, Yue Y, Zhang K, et al. A five-repeat micro-dystrophin gene ameliorated dystrophic phenotype in the severe DBA/2J-mdx model of Duchenne muscular dystrophy. Mol Ther Methods Clin Dev. 2017;6:216–30.
Zhang Y, Yue Y, Li L, Hakim CH, Zhang K, Thomas GD, et al. Dual AAV therapy ameliorates exercise-induced muscle injury and functional ischemia in murine models of Duchenne muscular dystrophy. Hum Mol Genet. 2013;22:3720–9.
Shin JH, Pan X, Hakim CH, Yang HT, Yue Y, Zhang K, et al. Microdystrophin ameliorates muscular dystrophy in the canine model of Duchenne muscular dystrophy. Mol Ther. 2013;21:750–7.
Yue Y, Pan X, Hakim CH, Kodippili K, Zhang K, Shin JH, et al. Safe and bodywide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno-associated virus. Hum Mol Genet. 2015;24:5880–90.
Kodippili K, Hakim C, Pan X, Yang HT, Yue Y, Zhang Y, et al. Dual AAV gene therapy for Duchenne muscular dystrophy with a 7-kb mini-dystrophin gene in the canine model. Hum Gene Ther. 2018;29:299–311.
Duan D. Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol Ther. 2018;26:2337–56.
Crawford GE, Faulkner JA, Crosbie RH, Campbell KP, Froehner SC, Chamberlain JS. Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. J Cell Biol. 2000;150:1399–410.
Rafael JA, Cox GA, Corrado K, Jung D, Campbell KP, Chamberlain JS. Forced expression of dystrophin deletion constructs reveals structure-function correlations. J Cell Biol. 1996;134:93–102.
Acknowledgements
We thank Dr. Glenn Morris, The Rober Jones and Agnes Hunt Orthopedic Hospital, Oswestry, Shropshire, United Kingdom, for providing us the antibody to detect dystrophin R17.
Funding
This work was supported by Duchenne Parent Project, The Netherlands (to Y.L.), and National Institute of Health (NS-90634), and Jackson Freel DMD Research Fund (to D.D.). The funding agencies are not involved in the study design, data collection, analysis and interpretation, and writing of the manuscript.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available from the corresponding authors on reasonable request.
Author information
Authors and Affiliations
Contributions
AP, JZ, YY, KZ, and YL conducted the experiments and acquired and analyzed the data. DD and YL designed the experiments and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
DD is a member of the scientific advisory board for Solid Biosciences and an equity holder of Solid Biosciences. The Duan lab and Lai lab has received research support from Solid Biosciences for unrelated projects.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. The full amino acid sequence of dystrophin R16/17-syntrophin PDZ.GFP.Pal fusion protein. The subdomains of the fusion protein was annotated by different colors (R16, R17, linker, syntrophin-PDZ, GFP, Pal). (PDF 56 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Patel, A., Zhao, J., Yue, Y. et al. Dystrophin R16/17-syntrophin PDZ fusion protein restores sarcolemmal nNOSμ. Skeletal Muscle 8, 36 (2018). https://doi.org/10.1186/s13395-018-0182-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13395-018-0182-x