
Abstract
Background
Better understanding of the signaling pathways that regulate human bone marrow stromal stem cell (hBMSC) differentiation into bone-forming osteoblasts is crucial for their clinical use in regenerative medicine. Chemical biology approaches using small molecules targeting specific signaling pathways are increasingly employed to manipulate stem cell differentiation fate.
Methods
We employed alkaline phosphatase activity and staining assays to assess osteoblast differentiation and Alizarin R staining to assess mineralized matrix formation of cultured hBMSCs. Changes in gene expression were assessed using an Agilent microarray platform, and data normalization and bioinformatics were performed using GeneSpring software. For in vivo ectopic bone formation experiments, hMSCs were mixed with hydroxyapatite–tricalcium phosphate granules and implanted subcutaneously into the dorsal surface of 8-week-old female nude mice. Hematoxylin and eosin staining and Sirius Red staining were used to detect bone formation in vivo.
Results
We identified several compounds which inhibited osteoblastic differentiation of hMSCs. In particular, we identified ruxolitinib (INCB018424) (3 μM), an inhibitor of JAK-STAT signaling that inhibited osteoblastic differentiation and matrix mineralization of hMSCs in vitro and reduced ectopic bone formation in vivo. Global gene expression profiling of ruxolitinib-treated cells identified 847 upregulated and 822 downregulated mRNA transcripts, compared to vehicle-treated control cells. Bioinformatic analysis revealed differential regulation of multiple genetic pathways, including TGFβ and insulin signaling, endochondral ossification, and focal adhesion.
Conclusions
We identified ruxolitinib as an important regulator of osteoblast differentiation of hMSCs. It is plausible that inhibition of osteoblast differentiation by ruxolitinib may represent a novel therapeutic strategy for the treatment of pathological conditions caused by accelerated osteoblast differentiation and mineralization.
Similar content being viewed by others


Background
Bone marrow stromal (also known as mesenchymal or skeletal) stem cells (BMSCs) exist within the bone marrow stromal and are capable for differentiation into mesoderm-type cells including bone-forming osteoblasts [1]. A number of signaling pathways have been implicated in regulating differentiation of human BMSCs (hBMSCs) into osteoblasts that include TGF-B [2], Wnt [3], and several intracellular kinases [4]. However, several other signaling pathways have been reported to regulate different aspects of stem cell biology in a number of stem cell systems [5] but their role in regulating hBMSC differentiation into osteoblastic cells are not well studied.
Chemical biology approaches using small molecules targeting specific intracellular or signaling factors are very important tools for studying stem cell differentiation and in vitro manipulation of stem cells (add ref). In addition, small molecules that induce stem cell differentiation are being employed as an alternative approach to classical stem cell differentiation protocols that require complex mixture of growth factors and cytokines, because of their scalable production, stability, ease of use, and low cost [6,7,8].
We have previously employed small molecule libraries to dissection mechanisms underlying differentiation potential of hBMSCs into osteoblasts [9] [4] and adipocytes [8].
Herein, we conducted an unbiased small molecule stem cell signaling library screen that covers several signaling pathways and identified ruxolitinib as an important regulator of osteoblast differentiation of hBMSCs.
Materials and methods
Stem cell signaling compound library
A stem cell signaling compound library, purchased from Selleckchem Inc. (Houston, TX, http://www.selleckchem.com) and consisted of 73 biologically active small molecular inhibitors, was employed in the presented study. An initial screen was conducted at a concentration of 3 μM.
Cell culture
We employed a telomerized hMSC line (hMSC-TERT) as a model for hBMSCs. The hMSC-TERT line was generated through an overexpression of the human telomerase reverse transcriptase gene (hTERT). hMSC-TERT exhibits the typical features of primary hMSCs including indefinite self-renewal and multipotency, in addition to the expression of all known markers of primary hMSCs [10,11,12]. The cells were maintained in DMEM, a basal medium supplemented with 4500 mg/L d-glucose, 4 mM l-glutamine, and 110 mg/L 10% sodium pyruvate, in addition to 10% fetal bovine serum (FBS), 1% penicillin–streptomycin, and 1% nonessential amino acids. All reagents were purchased from Thermo Fisher Scientific, Waltham, MA (http://www.thermofisher.com). Cells were incubated in 5% CO2 incubators at 37 °C and 95% humidity.
Osteoblast differentiation
The cells were cultured to 80–90% confluence and were incubated in osteoblast induction medium (DMEM containing 10% FBS, 1% penicillin–streptomycin, 50 μg/ml l-ascorbic acid (Wako Chemicals GmbH, Neuss, Germany, http://www.wako-chemicals. de/), 10 mM b-glycerophosphate (Sigma-Aldrich), 10 nM calcitriol (1a,25-dihydroxyvitamin D3; Sigma-Aldrich), and 10 nM dexamethasone (Sigma-Aldrich)). Each small molecule inhibitor was added at a concentration of 3 μM, in the osteoblast induction medium. The cells were exposed to the inhibitors throughout the differentiation period. Control cells were treated with osteoblast induction medium containing dimethyl sulfoxide (DMSO) as vehicle.
Cell viability assay
Cell viability assay was performed using alamarBlue assay according to the manufacturer’s recommendations (Thermo Fisher Scientific). In brief, cells were cultured in 96-well plates in 200 μl of the medium for 10 days, then 20 μl of alamarBlue substrate was added, and plates were incubated in the dark at 37 °C for 1 h. Readings were taken using fluorescent mode (Ex 530 nm/Em 590 nm) using a BioTek Synergy II microplate reader (BioTek Inc., Winooski, VT, USA).
Alkaline phosphatase activity quantification
To quantify alkaline phosphatase (ALP) activity, we employed the BioVision ALP activity colorimetric assay kit (BioVision, Inc., Milpitas, CA, http:// www.biovision.com/) with some modifications. The cells were cultured in 96-well plates. On day 10, the cells were rinsed once with phosphate-buffered saline (PBS) and fixed using 3.7% formaldehyde in 90% ethanol for 30 s at room temperature. Fixative was removed, and 50 μl of p-nitrophenyl phosphate solution was added to each well and incubated for 30–60 min. Optical densities were then measured at 405 nm using a SpectraMax/M5 fluorescence spectrophotometer plate reader. ALP enzymatic activity was normalized to cell number.
In vivo ectopic bone formation assay
All animal experimental procedures were approved by the Animal Care Committees of King Saud University. Cells were harvested via trypsinization, washed in PBS, and resuspended in culture medium with or without ruxolitinib. Approximately 5 × 105 cells were mixed with 40 mg of hydroxyapatite–tricalcium phosphate granules per each implant (HA/TCP, Zimmer Scandinavia, Albertslund, Denmark) and implanted subcutaneously into the dorsal surface of 8-week-old female nude mice, as previously described [13]. After 28 days, the implants were recovered, fixed in 4% paraformaldehyde, decalcified using formic acid solution (0.4 M formic acid and 0.5 M sodium formate), and embedded in paraffin. Tissue blocks were sectioned at 4 μm. Sections of paraffin-embedded implants were stained with hematoxylin and eosin and Sirius Red to identify areas of the formed bone.
Alkaline phosphatase staining
Cells were stained on day 10 of osteoblast differentiation. Cells cultured in 12-well plates were washed in PBS and fixed in 10 mM acetone/citrate buffer at pH 4.2 for 5 min at room temperature. The fixative was removed, and the Naphthol/Fast Red stain [0.2 mg/mL Naphthol AS-TR phosphate substrate (Sigma)] [0.417 mg/mL of Fast Red (Sigma)] was added for 1 h at room temperature. The cells were then rinsed with water and imaged under the microscope.
Alizarin Red S staining for mineralized matrix formation
Cells cultured in 12-well plates were stained on day 21 of osteoblast differentiation. The cells were washed twice with PBS and then fixed with 4% paraformaldehyde for 15 min at room temperature. Fixative was then removed, and the cells were washed with distilled water and stained with 2% Alizarin Red S Staining Kit (ScienceCell, Research Laboratories, Cat. No. 0223) for 20–30 min at room temperature. Subsequently, the dye was washed off with water and cells were imaged under the microscope.
RNA extraction and cDNA synthesis
Total RNA was isolated from cell pellets after 10 and 21 days of osteoblast differentiation using the total RNA Purification Kit (Norgen Biotek Corp., Thorold, ON, Canada, https://norgenbiotek.com/) according to the manufacturer’s protocol. The concentrations of total RNA were measured using NanoDrop 2000 (Thermo Fisher Scientific). cDNA was synthesized using 500 ng of total RNA and the Thermo Fisher Scientific High Capacity cDNA Transcription Kit according to the manufacturer’s protocol.
qRT-PCR
Expression levels of the mRNAs were validated using SYBR Green-based quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) with an Applied Biosystems ViiA™ 7 Real-Time PCR System (Thermo Fisher Scientific). Primers used in current study are listed in Table 1. The 2∆CT value method was used to calculate relative expression, and analysis was performed as previously described [14].
Gene expression profiling by microarray
One hundred fifty nanograms of total RNA was labeled using low input Quick Amp Labeling Kit (Agilent Technologies, Santa Clara, CA, http://www.agilent.com) and then hybridized to the Agilent Human SurePrint G3 Human GE 8 × 60 k microarray chip. All microarray experiments were performed at the Microarray Core Facility (Stem Cell Unit, Department of Anatomy, King Saud University College of Medicine, Riyadh, Saudi Arabia). The extracted data were normalized and analyzed using GeneSpring 13.0 software (Agilent Technologies). Pathway analyses were performed using the single experiment pathway analysis feature in GeneSpring 13.0 as described before [15]. Twofold cutoff and P (corr) < 0.05 (Benjamini–Hochberg multiple testing corrected) were used to determine significantly changed transcripts.
Statistical analysis
Statistical analysis and graphing were performed using Microsoft Excel 2010 and GraphPad Prism 6 software (GraphPad software, San Diego, CA, USA), respectively. Results were presented as mean ± SEM from at least two independent experiments. Unpaired t test was used to determine statistical significance and P values < 0.05 was considered statistically significant.
Results
Stem cell signaling library screen identified inhibitors of osteoblast differentiation of hBMSCs
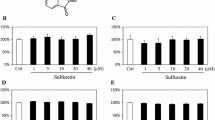
A stem cell signaling library consisting of 73 chemical compounds was used for the initial screen for their effects on osteoblastic differentiation of hBMSCs using ALP activity quantification as a read-out. All small molecules were tested at a concentration 3 μM. As shown in Fig. 1, the majority of small molecules reduced ALP activity of hBMSCs. Based on this initial screen, we chose 11 compounds (ruxolitinib (INCB018424), LY411575, BMS-833923, sotrastaurin, SB525334, LGK-974, ICG-001, BIO, TWS119, fasudil (HA-1077) HCl, and baricitinib (LY3009104, INCB028050)) for follow-up studies. The name of small molecule and their molecular targets are listed in Table 2. As shown in Fig. 2, several of the tested molecules inhibited osteoblastic differentiation of hBMSCs as evidenced by reduced ALP cytochemical staining at day 10 post-osteoblast differentiation induction (Fig. 2a) and this was concordant with the reduced ALP activity (Fig. 2b). These molecules did not exert significant effects on hBMSC viability (Fig. 2c). Among these small molecules, we chose ruxolitinib (INCB018424) for more detailed studies as it yielded the most consistent and potent effect on osteoblast differentiation and its effect on osteoblast differentiation of hBMSCs has not been studied before.

Functional screen of stem cell signaling small molecule library for their effects on osteoblast differentiation of human bone marrow stromal stem cells (hBMSCs). hBMSCs were induced into osteoblasts for 10 days in the presence of the indicated small molecule inhibitors (3.0 μM) or DMSO vehicle control. Data are presented as mean alkaline phosphatase (ALP) activity ± SEM, n ≥ 10 from three independent experiments. Small molecules are grouped according to their targeted signaling pathway. DMSO dimethyl sulfoxide. *P < 0.05; **P < 0.05; ***P < 0.0005

The effect of a selected panel of small molecules targeting multiple signaling pathways on osteoblast differentiation of hBMSCs. a Representative alkaline phosphatase (ALP) staining of hBMSCs on day 10 following treatment with the indicated compounds (concentration 3.0 μM). Images were taken at × 10 magnification using a Zeiss inverted microscope. b Quantification of ALP activity in hBMSCs following treatment with the indicated compounds (concentration 3 μM) versus vehicle-treated control cells at day 10. Data are presented as mean percentage ALP activity ± SEM, n > 16. **P < 0.05; ***P < 0.0005. c Cell viability assay using alamarBlue showing the relative cell viability in hBMSCs following treatment with the indicated compounds (3 μM) versus vehicle-treated control cells on day 10 post-osteoblast differentiation. Abbreviations: ALP alkaline phosphatase, DMSO dimethyl sulfoxide
Ruxolitinib inhibits mineralized matrix formation
To assess the effects of ruxolitinib on mineralized matrix formation, hBMSCs were treated with ruxolitinib (3 μM) and induced into osteoblast for 21 days. Alizarin Red staining demonstrated significant reduction in mineralized matrix formation in ruxolitinib-treated hBMSCs compared to vehicle-treated controls (Fig. 3a). Similarly, ruxolitinib reduced the expression of ALP and RUNX2 osteoblast gene markers measured on day 10 (b) or day 21 (c) post-osteoblast induction.

The effect of ruxolitinib on osteoblastic differentiation of hBMSCs. a hMSCs were induced into osteoblasts for 21 days in the absence (left panel) or presence (right panel) of ruxolitinib and were stained for mineralized matrix formation using Alizarin Red stain. Images were taken at × 10 magnification using a Zeiss inverted microscope. Quantitative RT-PCR analysis for gene expression of alkaline phosphatase (ALP) and RUNX2 in hBMSCs inducted into osteoblasts for 10 days (b) or 21 days (c) in the absence (blue) or presence (red) of ruxolitinib. Cells treated with DMSO were used as control. Gene expression was normalized to β-actin. Data are presented as mean fold change ± SEM (n = 6) from two independent experiments. ***P ≤ 0.0005. Abbreviations: ALP alkaline phosphatase, RUNX2 runt-related transcription factor 2, DMSO dimethyl sulfoxide
Ruxolitinib affects multiple signaling pathways during osteoblast differentiation of hBMSCs
To understand the molecular mechanism by which ruxolitinib inhibits osteoblast differentiation of hBMSCs, we performed global gene expression profiling and pathway analysis comparing ruxolitinib-treated and DMSO-treated control cells, during osteoblast differentiation. Figure 4a shows the hierarchical clustering based on the differentially expressed genes and demonstrates clear separation of ruxolitinib-treated and DMSO (vehicle)-treated control cells. We identified 847 upregulated and 822 downregulated genes (fold change ≥ 2.0; P (corr) < 0.05) (Additional file 1). Pathway analysis of the downregulated genes revealed strong enrichment for several cellular processes involved in osteoblast differentiation (e.g., TGFβ signaling, insulin signaling, endochondral ossification, and focal adhesion). A number of significantly enriched pathways in ruxolitinib-treated cells are illustrated as a pie chart (Fig. 4b), wherein the size of the slice corresponds to fold enrichment. Among the identified pathways, TGFβ signaling, insulin signaling, and focal adhesion signaling were prominent. These genetic pathways are known for their role in regulating osteoblast differentiation of hBMSCs. A number of genes from the enriched pathways (TNF, LIF, SOCS3, COMP, and THBS2) were selected and validated using qRT-PCR, which collectively corroborated the microarray data (Fig. 4c).

Ruxolitinib affects multiple pathways during osteoblastic differentiation of hBMSCs. a Heat map analysis and unsupervised hierarchical clustering performed on differentially expressed genes during osteoblast differentiation of ruxolitinib-treated compared to DMSO-treated control hBMSCs. b Pie chart illustrating the distribution of selected enriched pathway categories for the downregulated genes identified in osteoblast differentiated ruxolitinib-treated hBMSCs compared to DMSO-treated control cells. c Validation of a selected panel of downregulated genes during osteoblastic differentiation of ruxolitinib-treated hBMSCs compared to DMSO-treated control cells using qRT-PCR. Gene expression was normalized to β-actin. Data are presented as mean fold change ± SEM (n = 6) from two independent experiments; *P < 0.05; ***P < 0.0005
Effects of ruxolitinib on in vivo ectopic bone formation
To determine the regulatory role of ruxolitinib on in vivo bone formation, we implanted hBMSCs loaded on hydroxyapatite–tricalcium phosphate (HA/TCP) granules in the presence or absence of ruxolitinib into nude mice for 4 weeks. Histological analysis of the implants showed significant decrease in the formed ectopic bone in ruxolitinib-treated hBMSCs compared to control hBMSCs (Fig. 5a, b).

Ruxolitinib inhibits in vivo ectopic bone formation. Ruxolitinib-treated and control hBMSCs were implanted with hydroxyl apatite/tricalcium phosphate (HA/TCP) subcutaneously into NOD/SCID mice. The histology of in vivo bone formation was examined with H&E (a) and Sirius red (b) staining. Black arrows indicate the bone formation (× 20), and black line shows the bone formed zone with osteoblast between the HA and spindle-shaped hMSCs (× 40). Images were taken at × 20 (first row; scale bar = 100 μm) and × 40 (second row; scale bar = 50 μm) magnification using a light microscope. Abbreviation: H&E hematoxylin and eosin
Discussion
Small molecules, targeting specific signaling pathways, have recently emerged as a key tool to manipulate stem cell fate and differentiation potential in both mechanistic studies of stem cell biology as well as an approach to generate cells suitable for clinical use [6]. In the current study, we employed a well-characterized stem cell signaling library and performed unbiased functional screen on 73 small molecules targeting a number of signaling pathways relevant for hBMSC biology. These molecules covered a number of proteases, TGF-beta/Smad, JAK/STAT, Wnt, PI3K/Akt/mTOR, neuronal signaling, cell cycle, epigenetics, GPCR and G protein, Hedgehog, and GSK-3 inhibitors. Our initial screen identified several small molecule inhibitors, mainly targeting the JAK-STAT pathway, as potent inhibitors of osteoblastic differentiation of hBMSCs. In particular, ruxolitinib, a novel JAK-targeting small molecule inhibitor, was further studied and validated as a potent inhibitor of osteoblast differentiation.
JAK1 and JAK2 modulate the intracellular signaling of significant cytokines and growth factors for hematopoiesis and immune function including activation of signal transducers and activators of transcription (STAT). STAT3 is a ubiquitously expressed transcription factor activated by many cytokines and growth factors, including IL-6 family cytokines [16]. The receptors for the IL-6 family cytokines comprise of a ligand-binding subunit and a common signal-transducing subunit, gp130. Upon binding to their receptors, gp130 becomes activated leading to the activation of gp130-associated JAK (JAK1, JAK2, and TYK2) that subsequently lead to tyrosine phosphorylation of STAT3. Activated STAT3 localize to the nucleus and modulate various gene expression that regulate cell proliferation and differentiation in a cell-specific manner including bone metabolism [17]. The role of JAK-STAT signaling in osteoblastic differentiation is starting to unfold. For instance, inactivation and mutations of STAT3 in osteoblasts and osteocytes lead to distorted craniofacial and skeletal features, recurrent fractures, hyperextensible joints, reduce bone mass, strength, and load-driven bone formation, suggesting a role for STAT3 in osteoblast differentiation [18]. JAK-STAT signaling has been implicated in the maintenance of the stem cell pool in Drosophila and mammals [19]. Our findings from current study provide new evidence of potential involvement of JAK-STAT signaling in hBMSC biology.
Although in current study we did not investigate the downstream targets of ruxolitinib, it is well established that ruxolitinib is an ATP-competitive JAK1/2 inhibitor [20,21,22]. JAK1-deficient mice weighed less than their heterozygous and wild-type littermates, suggesting an important role for JAK1 in skeletal development [23]. Mouse embryonic fibroblasts derived from JAK2-deficient mice exhibited defects in signaling through a number of cytokine receptors, implying plausible role for JAK2 in skeletal development [24]. Cells treated with ruxolitinib exhibited diminished levels of phosphorylated STAT3, STA4, and STAT5 [25]. Therefore, it is plausible that ruxolitinib regulated osteoblastic differentiation of hBMSCs through inhibition of JAK-STAT3 signaling. Ruxolitinib might additionally inhibit other pathways known to be regulated by JAK, such as PI3K-AKT or ERK-JNK-p38, contributing to inhibition of osteoblastic differentiation [26, 27].
Global gene expression profiling of hMSC treated with ruxolitinib revealed multiple differentially regulated signaling pathways including TGFβ, insulin, endochondral ossification, and focal adhesion signaling, which are known to play an important role in regulating osteoblastic differentiation of hMSCs [28,29,30,31,32,33]. Those data are concordant with other published reports implicating protease-activated receptors [34], TGF-beta [2], Wnt/β-catenin [35], PI3K/Akt [36], cell cycle [37], and GPCR [38] signaling during osteogenesis.
Ruxolitinib is currently used in the clinic to treat patients with myelofibrosis, a clonal myeloproliferative neoplasm [39]. Ruxolitinib exhibited growth inhibition, apoptosis induction, and drop in inflammatory cytokine, mediated by inhibition of phosphorylate STAT via inhibition of JAK [20, 40]. No previous reports have been published regarding the effects of ruxolitinib on the osteoblast differentiation of hMSCs. We therefore investigated the expression of a selected panel of inflammatory cytokine (CXCL2, TNF, IL6, and CXCL1) from the microarray data during osteogenesis of hMSCs exposed to ruxolitinib and observed significant downregulation in the expression of those cytokines. While conceivable that small molecule inhibitors have specific targets, a number of studies have indicated deleterious effects of small molecule inhibitors on the biological function of mammalian cells [41, 42]. In particular, it was shown that hBMSCs are prone to cellular senescence under stress conditions [43]. Although we did not observe a significant change in cell viability of hBMSCs treated with ruxolitinib (3.0 μM) in the current study, it is plausible that inhibition of osteogenesis by ruxolitinib is in part due to possible effect on cellular senescence.
Enhancing bone formation and bone mass is needed in many conditions associated with bone loss such as in post-menopausal osteoporosis and glucocorticoid-induced osteoporosis, and suppression of osteogenic differentiation of hBMSCs by ruxolitinib may be relevant to a number of clinical conditions associated with ectopic bone formation or calcification including craniosynostosis and heart valve calcification [44, 45]. The clinical effectiveness of ruxolitinib in these conditions requires further studies.
Conclusion
Our unbiased small molecule screen identified ruxolitinib, a JAK-STAT inhibitor, as potent inhibitor of osteoblastic differentiation of hBMSCs. Inhibition of bone formation by ruxolitinib might represent a novel therapeutic strategy for the treatment of pathological conditions caused by accelerated osteoblast differentiation and mineralization.
Abbreviations
- ALP:
-
Alkaline phosphatase
- ALZR:
-
Alizarin Red
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- DMSO:
-
Dimethyl sulfoxide
- HA/TCP:
-
Hydroxyapatite–tricalcium phosphate
- hBMSCs:
-
Human bone marrow stromal stem cell
- hTERT:
-
Human telomerase reverse transcriptase
- JAK:
-
Janus kinase
- PBS:
-
Phosphate-buffered saline
- qRT-PCR:
-
Quantitative reverse transcriptase-polymerase chain reaction
References
Aldahmash A, Zaher W, Al-Nbaheen M, Kassem M. Human stromal (mesenchymal) stem cells: basic biology and current clinical use for tissue regeneration. Ann Saudi Med. 2012;32(1):68–77.
Elsafadi M, Manikandan M, Almalki S, Mobarak M, Atteya M, Iqbal Z, Hashmi JA, Shaheen S, Alajez N, Alfayez M, et al. TGFbeta1-induced differentiation of human bone marrow-derived MSCs is mediated by changes to the actin cytoskeleton. Stem Cells Int. 2018;2018:6913594.
Qiu W, Andersen TE, Bollerslev J, Mandrup S, Abdallah BM, Kassem M. Patients with high bone mass phenotype exhibit enhanced osteoblast differentiation and inhibition of adipogenesis of human mesenchymal stem cells. J Bone Miner Res. 2007;22(11):1720–31.
Jafari A, Siersbaek MS, Chen L, Qanie D, Zaher W, Abdallah BM, Kassem M. Pharmacological inhibition of protein kinase G1 enhances bone formation by human skeletal stem cells through activation of RhoA-Akt signaling. Stem Cells. 2015;33(7):2219–31.
Fakhry M, Hamade E, Badran B, Buchet R, Magne D. Molecular mechanisms of mesenchymal stem cell differentiation towards osteoblasts. World J Stem Cells. 2013;5(4):136–48.
Lu B, Atala A. Small molecules and small molecule drugs in regenerative medicine. Drug Discov Today. 2014;19(6):801–8.
Ding S, Schultz PG. A role for chemistry in stem cell biology. Nat Biotechnol. 2004;22(7):833–40.
Ali D, Hamam R, Alfayez M, Kassem M, Aldahmash A, Alajez NM. Epigenetic library screen identifies abexinostat as novel regulator of adipocytic and osteoblastic differentiation of human skeletal (mesenchymal) stem cells. Stem Cells Transl Med. 2016;5(8):1036–47.
Segar CE, Ogle ME, Botchwey EA. Regulation of angiogenesis and bone regeneration with natural and synthetic small molecules. Curr Pharm Des. 2013;19(19):3403–19.
Simonsen J, Rosada C, Sernici N, Justesen J, Stenderup K, Rattan S, Jensen T, Kassem M. Telomerase expression extends lifespan and prevents senescence-associated impairment of osteoblast functions. Nat Biotechnol. 2002;20(6):592–596.
Abdallah BM, Haack-Sorensen M, Burns JS, Elsnab B, Jakob F, Hokland P, Kassem M. Maintenance of differentiation potential of human bone marrow mesenchymal stem cells immortalized by human telomerase reverse transcriptase gene in despite of extensive proliferation. Biochem Biophys Res Commun. 2005;326(3):527–38.
Al-Nbaheen M, Vishnubalaji R, Ali D, Bouslimi A, Al-Jassir F, Megges M, Prigione A, Adjaye J, Kassem M, Aldahmash A. Human stromal (mesenchymal) stem cells from bone marrow, adipose tissue and skin exhibit differences in molecular phenotype and differentiation potential. Stem Cell Rev Rep. 2013;9(1):32–43.
Abdallah BM, Ditzel N, Kassem M. Assessment of bone formation capacity using in vivo transplantation assays: procedure and tissue analysis. Methods Mol Biol. 2008;455:89–100.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(−Delta Delta C) method. Methods. 2001;25(4):402–8.
Vishnubalaji R, Manikandan M, Fahad M, Hamam R, Alfayez M, Kassem M, Aldahmash A, Alajez NM. Molecular profiling of ALDH1(+) colorectal cancer stem cells reveals preferential activation of MAPK, FAK, and oxidative stress pro-survival signalling pathways. Oncotarget. 2018;9(17):13551–64.
Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, Akira S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A. 1997;94(8):3801–4.
Itoh S, Udagawa N, Takahashi N, Yoshitake F, Narita H, Ebisu S, Ishihara K. A critical role for interleukin-6 family-mediated Stat3 activation in osteoblast differentiation and bone formation. Bone. 2006;39(3):505–12.
Zhou H, Newnum AB, Martin JR, Li P, Nelson MT, Moh A, Fu XY, Yokota H, Li J. Osteoblast/osteocyte-specific inactivation of Stat3 decreases load-driven bone formation and accumulates reactive oxygen species. Bone. 2011;49(3):404–11.
Stine RR, Matunis EL. JAK-STAT signaling in stem cells. Adv Exp Med Biol. 2013;786:247–67.
Ostojic A, Vrhovac R, Verstovsek S. Ruxolitinib: a new JAK1/2 inhibitor that offers promising options for treatment of myelofibrosis. Future Oncol. 2011;7(9):1035–43.
Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, Caulder E, Wen X, Li Y, Waeltz P, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15):3109–17.
Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, Estrov Z, Fridman JS, Bradley EC, Erickson-Viitanen S, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–27.
Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, King KL, Sheehan KC, Yin L, Pennica D, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. 1998;93(3):373–83.
Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, Vanin EF, Bodner S, Colamonici OR, van Deursen JM, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93(3):385–95.
Bottos A, Gotthardt D, Gill JW, Gattelli A, Frei A, Tzankov A, Sexl V, Wodnar-Filipowicz A, Hynes NE. Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat Commun. 2016;7:12258.
Abell K, Watson CJ. The Jak/Stat pathway: a novel way to regulate PI3K activity. Cell Cycle. 2005;4(7):897–900.
Winston LA, Hunter T. Intracellular signalling: putting JAKs on the kinase MAP. Curr Biol. 1996;6(6):668–71.
Akhurst RJ, Fitzpatrick DR, Fowlis DJ, Gatherer D, Millan FA, Slager H. The role of TGF-beta-S in mammalian development and neoplasia. Mol Reprod Dev. 1992;32(2):127–35.
Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370(6488):341–7.
Lehnert SA, Akhurst RJ. Embryonic expression pattern of TGF-beta type-1 RNA suggests both paracrine and autocrine mechanisms of action. Development. 1988;104(2):263–73.
Pelton RW, Hogan BLM, Miller DA, Moses HL. Differential expression of genes encoding TGFs beta-1, beta-2, and beta-3 during murine palate formation. Dev Biol. 1990;141(2):456–60.
Salasznyk RM, Klees RF, Williams WA, Boskey A, Plopper GE. Focal adhesion kinase signaling pathways regulate the osteogenic differentiation of human mesenchymal stem cells. Exp Cell Res. 2007;313(1):22–37.
Hayrapetyan A, Jansen JA, van den Beucken JJJP. Signaling pathways involved in osteogenesis and their application for bone regenerative medicine. Tissue Eng Part B-Reviews. 2015;21(1):75–87.
Georgy SR, Pagel CN, Ghasem-Zadeh A, Zebaze RM, Pike RN, Sims NA, Mackie EJ. Proteinase-activated receptor-2 is required for normal osteoblast and osteoclast differentiation during skeletal growth and repair. Bone. 2012;50(3):704–12.
Saidak Z, Le Henaff C, Azzi S, Marty C, Da Nascimento S, Sonnet P, Marie PJ. Wnt/beta-catenin signaling mediates osteoblast differentiation triggered by peptide-induced alpha5beta1 integrin priming in mesenchymal skeletal cells. J Biol Chem. 2015;290(11):6903–12.
Fujita T, Azuma Y, Fukuyama R, Hattori Y, Yoshida C, Koida M, Ogita K, Komori T. Runx2 induces osteoblast and chondrocyte differentiation and enhances their migration by coupling with PI3K-Akt signaling. J Cell Biol. 2004;166(1):85–95.
Chen L, Holmstrom K, Qiu W, Ditzel N, Shi K, Hokland L, Kassem M. MicroRNA-34a inhibits osteoblast differentiation and in vivo bone formation of human stromal stem cells. Stem Cells. 2014;32(4):902–12.
Luo J, Zhou W, Zhou X, Li D, Weng J, Yi Z, Cho SG, Li C, Yi T, Wu X, et al. Regulation of bone formation and remodeling by G-protein-coupled receptor 48. Development. 2009;136(16):2747–56.
Randhawa J, Ostojic A, Vrhovac R, Atallah E, Verstovsek S. Splenomegaly in myelofibrosis--new options for therapy and the therapeutic potential of Janus kinase 2 inhibitors. J Hematol Oncol. 2012;5:43.
Zacharaki D, Ghazanfari R, Li H, Lim HC, Scheding S. Effects of JAK1/2 inhibition on bone marrow stromal cells of myeloproliferative neoplasm (MPN) patients and healthy individuals. Eur J Haematol. 2018;101(1):57–67.
Weiss WA, Taylor SS, Shokat KM. Recognizing and exploiting differences between RNAi and small-molecule inhibitors. Nat Chem Biol. 2007;3(12):739–44.
Zhang Y, Li W, Laurent T, Ding S. Small molecules, big roles -- the chemical manipulation of stem cell fate and somatic cell reprogramming. J Cell Sci. 2012;125(Pt 23):5609–20.
Alessio N, Del Gaudio S, Capasso S, Di Bernardo G, Cappabianca S, Cipollaro M, Peluso G, Galderisi U. Low dose radiation induced senescence of human mesenchymal stromal cells and impaired the autophagy process. Oncotarget. 2015;6(10):8155–66.
Augello A, De Bari C. The regulation of differentiation in mesenchymal stem cells. Hum Gene Ther. 2010;21(10):1226–38.
Schoen FJ, Levy RJ. Calcification of tissue heart valve substitutes: progress toward understanding and prevention. Ann Thorac Surg. 2005;79(3):1072–80.
Acknowledgements
We would like to thank the Deanship of Scientific Research at King Saud University (Research Group No. RG-1438-033) for funding this work.
Funding
This work was supported by the Deanship of Scientific Research at King Saud University Research Group No. RG-1438-033.
Availability of data and materials
Data are available upon request.
Author information
Authors and Affiliations
Contributions
NA performed the experiments and participated in the manuscript writing. AA, RV, MM, MA, and AS performed the experiments. MA and AA were involved in the conception and design. MK was involved in the conception and design and in manuscript editing. NMA obtained the funding, conceived the study, and finalized the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal experiments received the appropriate ethical approval from the King Saud University Ethical Research Committee.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
List of differentially expressed genes (2.0 FC, p corr < 0.05) in human bone marrow mesenchymal stem cells (hBMSCs) differentiated into osteoblasts (day 10) in the presence of Ruxolitinib compared to DMSO. Differentially expressed genes (2.0 FC, p corr < 0.05) in human bone marrow mesenchymal stem cells (hBMSCs) differentiated into osteoblasts (day 10) in the presence of Ruxolitinib compared to DMSO detected using microarray. (XLSX 365 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
AlMuraikhi, N., Ali, D., Alshanwani, A. et al. Stem cell library screen identified ruxolitinib as regulator of osteoblastic differentiation of human skeletal stem cells. Stem Cell Res Ther 9, 319 (2018). https://doi.org/10.1186/s13287-018-1068-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-018-1068-x