
Abstract
Patients diagnosed with chromosome microdeletions or duplications, known as copy number variants (CNVs), present a unique opportunity to investigate the relationship between patient genotype and cell phenotype. CNVs have high genetic penetrance and give a good correlation between gene locus and patient clinical phenotype. This is especially effective for the study of patients with neurodevelopmental disorders (NDD), including those falling within the autism spectrum disorders (ASD). A key question is whether this correlation between genetics and clinical presentation at the level of the patient can be translated to the cell phenotypes arising from the neurodevelopment of patient induced pluripotent stem cells (iPSCs).
Here, we examine how iPSCs derived from ASD patients with an associated CNV inform our understanding of the genetic and biological mechanisms underlying the aetiology of ASD. We consider selection of genetically characterised patient iPSCs; use of appropriate control lines; aspects of human neurocellular biology that can capture in vitro the patient clinical phenotype; and current limitations of patient iPSC-based studies. Finally, we consider how future research may be enhanced to maximise the utility of CNV patients for research of pathological mechanisms or therapeutic targets.
Similar content being viewed by others


Background
Autism spectrum disorders (ASD) are a complex and heterogeneous group within the wider spectrum of neurodevelopmental disorders (NDD), which in its entirety also encompass intellectual disability (ID), attention-deficit/hyperactivity disorders (ADHD) and schizophrenia. ASD in particular are characterised by deficits in social interaction, communication difficulty and the presence of restricted, repetitive and stereotyped patterns of behaviour [1]. However, individual autistic patients often have features of other NDD, as well as comorbidities, such as epilepsy and anxiety [2]. As a consequence, their study therefore can provide insights across a range of common psychiatric disorders.
As seen with other NDD, the underlying biological origins of ASD begin during brain development and neural maturation; however, the clinical symptoms only emerge progressively during infancy, and as a consequence it is often difficult to diagnose at early post-natal stages. In addition, the spectrum of symptoms and phenotypes vary considerably from one individual to the next, presenting a challenge to identify those changes that directly arise from early developmental deficits and those that accumulate either indirectly or due to the effects of external factors. These complications hinder identification of the basic pathophysiological mechanisms that lead to ASD and hence hamper development of effective therapies.
Molecular and cellular analysis of human patients is generally prospective with data mostly derived from post-mortem tissue. As mentioned above, such studies are subject to the confounds of secondary effects and record the outcomes of underlying disease mechanism rather than directly probe the causative mechanisms. Animal models can be highly informative for the study of a basic mechanism; however, it is difficult to directly translate between observed patient phenotype and animal models. A particular weakness is the ability to capture the phenotypic variation across the patient population.
Human stem cell models offer an opportunity to directly study the molecular and cellular mechanisms of diseases. Key to this approach is the generation of human-induced pluripotent stem cells (iPSCs) derived from patient cells. These are generated by reprogramming of somatic cells into pluripotent stem cells from which many cell types can be differentiated, including neurons and glial cells. Importantly, they can be easily obtained in the clinic from fibroblasts (skin biopsies), keratinocytes (hair roots) [3], T lymphocytes (peripheral blood) [4, 5] and exfoliated renal epithelial cells from urine samples [6, 7]. Importantly, patient iPSCs enable the in vitro study of different cells types in isolation or co-culture in order to investigate cell function. Uniquely they can track the development profile of patient cell differentiation. More recently the capacity of iPSCs to form 3D organoids has opened up the possibility to investigate the interaction of multiple cell types in a more brain-like microenvironment. Methods for increasing reproducibility of brain organoid differentiation are improving substantially [8, 9] and being exploited to mechanistically dissect the effect of genetic lesions causing ASD and ID [10,11,12], as well as the role of specific genes and molecular modules key to human-specific neuronal differentiation trajectories and pathophysiology [13].
The major question is how to identify the relevant cellular phenotypes that converge on the common pathophysiological mechanisms underlying patient aetiology. Recent technical advances, particularly the advent of microarray technologies and whole-genome sequencing (WGS), have heralded a new era for detection of genetic risk loci for ASD [14, 15]. Unfortunately, most genetic risk for ASD is due to variations on 100’s of loci dispersed across the genome, each contributing only a small component to the overall level of genetic risk. Over the last decade, an accumulation of genetic evidence has pointed to three broad aspects of neuronal cell biology associated with elevated risk: synapse biology, gene regulation and neuro-inflammatory pathways. Emerging studies indicate that this dysregulated cell physiology contributes to circuit dysfunction, cortical layer malformation and white matter alteration seen within the patient brain. All of these biological processes and functional pathways can be investigated in patient iPSCs, offering capabilities beyond studies of post-mortem tissue or preclinical rodent models.
The dispersed nature of common genetic risk creates difficulties for cell modelling with most patient iPSCs. Although common genetic risk can be calculated as an overall polygenic risk score (PRS), this does not provide a direct concordance between specific changes at a genetic locus and the symptoms associated with ASD. In contrast, single nucleotide variants (SNVs) and copy number variants (CNVs) have much higher genetic penetrance for risk of developing ASD and other NDD [16] and hence changes at single or a few loci may make a major contribution to the clinical phenotype. Both SNVs and CNVs are a lot less common within the general patient population, but at present common clinical availability of array comparative genomic hybridisation (aCGH) technology makes CNVs more likely to be identified by cytogenomic screening. Accordingly, patients harbouring pathogenic CNVs present a powerful opportunity to relate genetic risk to patient clinical presentation. Here, we examine how iPSC-based studies using CNV patients can provide insights into the relationship of risk genetics to biological outcomes and can be utilised for the elucidation of disease mechanism.
Main text
An estimated 5–10% of all ASD cases carry CNVs [14], compared to 1.4–2.5% of all schizophrenia cases [15, 17]. Often CNVs arise spontaneously or de novo, although they can also be inherited in families, and at least 90 pathogenic CNVs have been reported for ASD [18]. There is however a core of CNVs associated with strong evidence of association with ASD and prevalent amongst people referred for genetic testing (Table 1) [19,20,21,22]. It is clear that despite having high genetic penetrance, CNVs present with considerable clinical variation in severity, phenotypic profile and co-morbidity, even between individuals with CNVs at the same chromosomal locus. For example, it is well established that a deletion at the 22q11.2 locus (22q11.2DS) is associated with ASD, but when children were identified first by genetic screening as few as 16% had ASD using stringent assessment criteria, although more than 80% showed diagnostic criteria for at least one psychiatric disorder and approximately 60% exhibited characteristics of ADHD [23]. In adults, 25% of patients with 22q11.2DS develop schizophrenia, 70% of the individuals possessing the same size deletion at the 22q11.2 locus exhibit congenital heart conditions [24,25,26].
These observations define the key questions of both genetics and biology that CNV patient iPSCs can address by the development of cell modelling of ASD and other NDD. In this review, we will discuss how these studies help understand the mechanisms underlying the genotype-to-phenotype relationship for ASD risk; what aspects of ASD can be meaningfully modelled in iPSC-derived neurons, and what limitations these studies possess. Finally, we will consider what methodological approaches are required to advance these studies.
Genotype-to-phenotype relationship in NDD patients harbouring pathogenic CNVs
The possible reasons for variation in the relationship between genotype and phenotype for different patients are still uncertain; however, they present an important consideration when choosing which patients to select for further study. What additional genetic factors and genomic mechanisms might increase phenotypic variation of patients with apparent similar risk loci?
First, the size, and hence number of genes affected at individual loci can vary considerably, with many of the larger CNVs having a number of different break points. The 22q11.2 locus for example can occur between two of four different breakpoints (A–D) with five different forms of deletion reported in the patient population [24]. In addition, break point regions can have complex local sequence changes, such as short sequence inversions or insertions and deletions (indels) that vary from one individual to the next but are not detected by the commonly used sequencing technologies and arrays [27]. With the advent of a new generation of single-molecule real-time (SMRT) sequence technologies that are capable of very long reads in each run, this level of variation is likely to be resolved in the future. Furthermore, it is also becoming very clear that the 3D chromatin structure, such as chromosome loops (topological associated domains, TADs) and long-range chromatin interactions, also play an important role in gene regulation. CNVs may influence or even disrupt gene regulation beyond the specific sequences contained within them [28]. Again, techniques are becoming available to accurately map these changes. Although these sources of genome variation are still a major challenge for CNV analysis, they also present a considerable opportunity for iPSC-based studies due to their ability to draw together genomics, transcriptomics and quantitative cell phenotyping.
A second major source of genomic variation arises due to patient diagnosis. As in the great majority of published studies patients are initially selected on the basis of their clinical presentation, for example congenital abnormality or developmental delay, which leads to then being referred for genetic testing. As a consequence, there is likely to be acquisition bias and under-representation of those individuals who have little or no pathology associated with their CNV. This bias may select for patients who possess additional background variation in their genome that enhances the effect of a CNV but is not detected by standard clinical screening. Studies on the impact of carrying several “pathogenic” CNVs indicate that children who carried two large CNVs of unknown clinical significance were eight times as likely to have developmental delay than patients with a single CNV [29]. Observations of the 16p12.1 deletion suggest a two-hit model for severity of its associated impact on developmental delay [30]. Possession of common risk variants in an individual background genome may also influence severity and clinical presentation. A study on schizophrenia patients showed higher PRS in individuals with low-penetrant CNVs in comparison to those with high-penetrant CNVs [31] and children with ADHD showed lower PRS when carrying large, rare CNVs in comparison to children with ADHD without such CNVs [32]. These results support the proposition that CNVs exert the same genetic pressure on risk as common variants but in general are more penetrant.
Finally, when using patient iPSCs for modelling it is important to take into account that for some CNVs, ASD is 4-fold less prevalent in females than males, yet there is an excess of deleterious CNVs in the female population, suggesting the existence of a protective effect [33]. It is currently unclear whether this protection extends in cells derived from female patients after reprogramming and/or during in vitro neuro-differentiation but needs to be considered as a possible confound when comparing between cell lines from different patients and non-patient controls.
Appropriate controls for patient iPSC studies
The genetic observations above underlie the need for careful selection of controls for iPSC studies. In an ideal situation, iPSCs derived from parents and siblings should be used to generate control cells alongside patient cells. In addition, genome editing techniques address the need for controls by standardising genetic backgrounds via generation of isogenic cell lines. Here, CRISPR/Cas9-mediated genome editing can be used to target a single gene within a CNV, so that the edited iPSC lines will have an identical genetic background to the parental line, minimising heterogeneity and phenotypic variability arising due to the genetic differences in the genomic background [34]. Multiple rounds of CRISPR can be used to sequentially disrupt more than one gene to model aspects of larger CNVs, or pairs of gRNA used to generate large genomic deletions and other rearrangements to create cell models with up to 1Mbp deletions [35] or reciprocal CNVs in human iPSCs [36]. In all cases, care should be taken to avoid introduction of off-target mutations leading to the small indels or even CNVs elsewhere in the genome. As a minimum, it is advisable to study multiple, independent engineered cells lines and genotype each using array screening. In future, availability and prevalence of WGS technologies may allow for more in-depth analysis. Finally, ideally engineered cells lines can be “rescued” by further engineering or reversible transgenesis using a piggyBac transposon system [37] to the original gene copy number.
What aspects of ASD can be modelled in iPSCs derived from CNV patients?
Although not without the challenges described above, a high degree of genetic penetrance makes a strong biological case for use of iPSCs from patients harbouring pathogenic CNVs as the basis for creation of disease-relevant cell assays. However, beyond a simple justification of providing access to human cell physiology, patient iPSC studies need to be tailored to align with the underlying biology observed for ASD and other NDD. Table 2 (and Supplementary Tables 2a and 2b) list those iPSC lines and their analysis that have been reported to date.
All NDD conditions are characterised by aberrant brain and cognitive function that arises during pre-natal or early post-natal life stages. Ultimately, these changes result in altered neuronal function and are strongly associated with synapse function and neuronal activity. However, genetic risk does not necessarily arise directly from mutation of synaptic protein-encoding genes and may also arise from deficits that occur in early neurodevelopment leading to abnormal neurogenesis or synaptogenesis. In the following section, we relate the aspects of ASD biology to the different modes of study available for patient iPSCs.
Synapse biology and neuronal activity
Accumulating evidence indicates altered brain connectivity as a common feature across all psychiatric disorders, implying underlying abnormalities of the brain circuitry [61]. Analysis of neuronal connectivity via synapses in cultured neurons can be performed using immunocytochemistry of synaptic proteins to examine synaptic density, rabies virus techniques to identify which neurons are connected via synapses, and a range of electrophysiological techniques that allow the detection of postsynaptic currents and potentials [62].
Studies on the glutamatergic excitatory neuronal synapse from ASD patient iPSCs with a 22q13.3 deletion or de novo mutations of the synaptic protein Shank3, which is located within the CNV, showed that these neurons have significantly reduced numbers of synapses and a corresponding decrease in synaptic transmission. These deficits can be reversed by treatment with the insulin-related hormone IGF-1 [55, 63]. Consistent with synaptic transmission deficits, iPSC-derived neurons from 15q11-q13 duplication syndrome were shown to be associated with delayed action potential maturation and increased synaptic event frequency. iPSC-derived neurons from 16p11.2 deletion and duplication were again associated with a decreased number of synapses. However, in contrast to other reports, the amplitude of excitatory postsynaptic currents of 16p11.2-derived neurons was significantly higher suggesting that they have increased synaptic strength [52]. Investigation of patient iPSCs with the 9q34 del (Kleefstra Syndrome) indicated a network disruption that correlates with increased expression of the NMDA subunit GluN1 that can be reversed by the NMDA blocker MK-801 [44].
These observations suggest that altered brain activity associated with CNVs can be captured in vitro in iPSC culture, offering an excellent platform for pharmacology of human neuronal systems. However, they are not the only differences that can be observed using patient iPSCs.
Gene regulation and neurodevelopment
iPSCs uniquely offer the means to follow neurodevelopment in vitro at the cellular level, providing a direct way to follow timing and fate during cell differentiation. This can be readily investigated by monitoring gene expression by RNA profiling techniques, such as RNA-seq, and assayed in conjunction with immunocytochemistry and cellomic approaches to localise changes to specific cells.
Studies of iPSCs derived from idiopathic ASD patients with no genetic or clinical stratification identified gene modules (sets of co-expressed genes), which are mis-regulated in the patient lines [64]. Affected modules, included those involved in synaptic transmission, correlated with altered neuronal network activity, as measured by multi-electrode array (MEA) and calcium signalling. A recent iPSC study focused on ASD patients who exhibit macrocephaly identified heterochronic disease-associated changes of gene expression and chromatin accessibility during their neuronal differentiation that could be reversed via expression of the neurogenic transcription factor NGN2 [65].
Direct examples of a gene regulatory change due to a CNV are Kleefstra syndrome and 7q11.23 (WBS). Loci of both CNVs contain epigenetic regulators that repress transcription to ultimately lead to synaptic dysfunction, either through loss of the EHMT1 gene, which generates the suppressive histone methylation H3K9me2 [66] in the case of Kleefstra syndrome or GTF21 in WBS [68]. GTF21 dosage imbalance is responsible for a large proportion of the transcriptional deregulation in WBS acting mainly as a transcriptional activator or cooperating with the H3K4 demethylase LSD1 in mediating transcriptional repression [43, 67]. The CNV 1q21.1 also contains a remodeller but its gene regulatory function has yet to be investigated. In addition to direct changes where CNV result in loss of a gene regulator, indirect changes can also lead to altered expression, for example the transcriptional profile changes associated with loss of Shank3 [68]. RNA profiling methods and cell localisation approaches can be combined together using single-cell RNA-seq to observe altered expression as cells diverge into different cell states, as reported from WBS and NRXN-1 CNVs [40, 67].
Neuro-inflammatory pathways
A third major biological pathway identified from GWAS is neuro-inflammation. Although this seems quite distinct from synaptic biology and neurodevelopment, in the context of the nervous system immunological pathways play a major part in shaping neuronal interactions and function. Elevated levels of TNF-α, IL-1β, IL-6 and IL-17 in the brains of ASD patients support the notion that dysregulated immunomodulatory pathways contribute towards pathology [69].
Post-mortem brain tissue from ASD patient has shown an increased microglial activation in the prefrontal cortex and microglia-specific gene expression has been found to be altered. Deficits in microglial activity were shown to reduce synaptic pruning leading to altered brain connectivity due to an accumulation of immature synapses [70]. It is not yet clear whether the increase in peripheral inflammation reported for 22q11, 16p11.2, 3q and 7q11.23 CNVs are reflected in microglial dysfunction, but loss of the Cyfip1 gene present within the 15q11.2 CNV in mice leads to increased neurogenesis due to a failure of microglial-induced neuronal apoptosis [71]. Protocols for differentiation of microglia from iPSCs have been developed, and these microglia were shown to secrete pro-inflammatory cytokines upon stimulation. However, their effect on synaptic pruning and brain development has not been yet reported.
Although neuronal dysfunction receives most attention, iPSC-based studies have indicated dysfunction of glial cells. In neuron-astrocyte co-cultures, it was shown that ASD patient astrocytes can interfere with neuronal development, while control astrocytes could rescue changes in neuronal morphology and synaptogenesis in non-syndromic ASD cultures [72]. Increased levels of IL-6 in the ASD astrocytes were suggested to underlie astrocyte actions, in line with earlier studies showing involvement of immune system dysregulation in ASD [73]. Recent single-cell expression studies on iPSC-derived neural stem cell cultures also revealed that NRXN1-α deletions shift neural cell development into higher proportions of astroglial cells [40], thereby affecting functional maturation of neurons. In addition to astroglia, a recent study also indicated involvement of oligodendroglia in neuronal phenotypes associated with ASD, as shown by neuron-oligodendrocytes co-cultures generated from tuberous sclerosis complex (TSC) patient cells [74]. Whether the cellular changes in morphology are also linked to the ASD phenotypes is so far unclear but will continue to be an expanding area for CNV patient iPSC studies.
Cortical abnormalities
It has been widely reported that brains of children with ASD have accelerated growth compared to non-patient controls of similar age, known as macrocephaly [75,76,77,78]. Magnetic resonance imaging (MRI) studies indicate wide-reaching phenotypic impact in some rare CNVs leading to substantial size and shape changes in the brain, e.g. 22q11 [79, 80], 7q11.23 deletion [81, 82], 15q11.2 [83,84,85] and 16p11.2 proximal [86,87,88] and distal CNVs [89], potentially underlining a neurodevelopmental component to the disorders. As such, a recent study using iPSCs from ASD patients with macrocephaly identified disease-associated changes at transcriptome and cellular levels that are present in a very early stage of neural stem cells and established an ASD specific gene signature. More importantly, overexpression of module-specific gene signatures in healthy cells was enough to recapitulate ASD disease-specific cellular phenotypes [65]. These findings demonstrate that there are key nodes within dysregulated gene networks that are related to ASD and may represent promising targets for therapeutic intervention.
The presence of additional numbers of neurons in the prefrontal cortex of the post-mortem brain from ASD patients complements clinical observations [75]. Furthermore, changes in neuronal density, neurogenesis and increased cortical thickness were found in subjects diagnosed with ASD [90, 91]. Using iPSCs, Deshpande et al. [52] assessed changes in brain growth phenotypes, such as cellular morphology in iPSC-based cortical cultures derived from 16p11.2 deletion and duplication patients. In line with clinical changes, neuronal cultures developed from 16p11.2 deletion iPSCs possessed increased soma size and dendritic length, while those from the duplication had reduced cell size and dendritic length. A further correlation between brain size and increased proliferation of neuronal progenitors has also been shown in iPSC-based cultures of individuals with idiopathic ASD [92].
Use of 3D brain organoid cultures have shown that changes in even a single gene, such as FOXG1 [46] and CHD8 [12], can lead to dysregulation of the cortical layer formation, cell migration and cell division and potentially increased differentiation GABAergic neurons. Organoid modelling 17p13.3 deletion (Miller-Dieker syndrome) demonstrated mitotic defects in the outer radial glia, cell migration and also an overproduction of GABAergic neurons [10, 54]. However, although the 17p13.3 deletion is associated with both ID and unprovoked seizures, it is the duplication that is associated with risk of ASD, and this has not been reported to have a deficit in GABAergic neuronal number. Furthermore, there is no consensus on whether ASD is associated with decrease or increased GABAergic neurons, even whether it is an excitatory/inhibitory imbalance per se, not its direction, that increase risk [93]. Organoids derived from CNV-associated patient offer good potential to resolve this question in future.
White matter alterations
Emerging studies suggest that white matter pathology also contributes towards the pathogenesis of ASD. Patients show regional decrease of white matter and increase of the grey matter [94,95,96]. The presence of supernumerary neurons within the white matter of the subcortical region is a commonly observed anomaly of ASD. Additionally, white matter integrity as measured by fractional anisotropy seems to be altered in 22q11.2 deletion patients [97] and 16p11.2 deletion is also associated with decreased myelination in the subcortical region of the brain [98]. A recent animal study of Cyfip1 function in the corpus callosum showed that its deletion decreases the myelinating potential of oligodendrocytes [99]. Although it is still not understood what induces hypomyelination in the ASD, this may arise from deficits in oligodendrocyte progenitor cell differentiation or dysregulated apoptosis of mature oligodendrocytes leading to decreased neuronal myelination. This interaction can be modelled with patient iPSCs, and cells from ASD patients with loss of function TSC-1 or -2 mutations exhibit oligodendrocyte-neuronal signalling and altered cell differentiation [74]. Given the evidence from brain imaging and animal studies, future in vitro cell studies of iPSCs derived from patients harbouring pathogenic CNVs are likely to be very informative.
Limitations
While the “What aspects of ASD can be modelled in iPSCs derived from CNV patients?” section outlines how in vitro cell cultures of iPSCs derived from patients harbouring pathogenic CNVs can capture disease-relevant features seen in the patient brain, we have to recognise a number of limitations and potential confounds.
-
1.
Cellular reprograming can contribute to formation of de novo CNVs in pluripotent stem cells during early cell passage, although both the size and total number seems to decrease with subsequent passages possibly due to negative selection [100]. Such genomic instability presents a potential serious confound for studies of iPSCs derived from patients harbouring pathogenic CNVs and has been shown to have adverse effect on pluripotency, cell proliferation and differentiation [101,102,103,104], all features of the patient cell phenotype. Careful monitoring of the cell karyotype and regular array-based or WGS genotyping is required to eliminate any cell lines that have accumulated artefactual CNVs during their generation. It is also recommended that newly generated lines should be grown for a minimum of 10 passages to ensure genetic stability, coupled with genotyping and differentiation.
-
2.
Gene expression analyses indicate that iPSC-derived neurons appear to be in a pre-natal state [105, 106] and several ageing-associated changes are erased during the reprogramming process [107]. This is a useful feature when using iPSCs for studying neurodevelopmental deficits. It does however mean that genome imprinting and other epigenetic changes associated with parental and environment interactions may be lost [108]. Future studies modelling ASD may benefit from monitoring changes in chromatin modification at imprinted and other loci.
-
3.
Differentiation of human iPSCs extends over long culture times, taking up to 100 days or more to fully form functionally mature neurons, and even then, they often show characteristics of foetal embryonic neurons [109]. This prolonged process affords plenty of opportunities for small variations in culture conditions to introduce differences from one culture to the next. Direct comparisons of gene expression, by use of RT-qPCR or RNA-seq, and cell electrophysiology need to be made to ensure that cultures have reached the same level of maturity. Currently, whole-cell recordings of iPSC-derived neurons are routinely performed and provide accurate measurements of the intrinsic properties of these cells. Relevant parameters to study the degree of neuronal differentiation (such as stable resting membrane potential, input resistance, membrane capacitance and action potential characteristics) can be determined by measuring the voltage response of the cell to injected hyperpolarizing or depolarizing current pulses. Where fine comparisons are needed between patient and non-patient controls for different cell types, single-cell RNA-seq or even Patch-seq for neurons, which combines patch-clamp and RNA-seq [110], may be beneficial.
-
4.
Finally, the in vitro culture conditions, whether 2D or 3D, employed for iPSC studies rely on the self-assembly of cell-cell contacts and synaptogenesis. Although this may model the dynamics of neurogenesis in the brain, it does not recapitulate the higher order organisation and circuitry seen in the human brain. This does currently limit the scope of iPSC work, but solutions are now in development, such as MEA analysis of 3D brain organoids [111].
Future development
Current usage of iPSCs from ASD and other NDD patients with associated CNV is improving our understanding of disease mechanism at the neural cell level. Importantly as discussed in the “What aspects of ASD can be modelled in iPSCs derived from CNV patients?” section, the cell phenotypic data that can be gained from these studies do align with observations from patients and in the clinic. This demonstrates the utility of iPSCs derived from patients harbouring pathogenic CNVs. But how can this be developed further to enhance their future role for study of the origins of ASD and ultimately for development of new therapeutics?
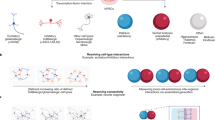
We propose that the next generation of iPSC studies need to expand in three domains: genomic complexity, cell assay complexity and scale (Fig. 1). In combination, all three will build an enhanced cell-based platform for the study of ASD and other NDD in vitro and at the cellular level with increasing construct validity.

Three domains for future expansion of iPSC studies. Increases in the number of patient iPSCs within a study (from low to high); assay complexity (from single parameter of 2D cultures to complex, multi-parameteric, high content assays on co-cultures or in 3D structured and organoid conditions) and increases in cell genetic complexity (from single CNV (monogenic) to multiple CNV and increasing polygenic (PRS) genomic background)
Genomic complexity
In the simplest cases, CNV patients, in which only a single gene is deleted or duplicated and also exhibits high genetic penetrance, can approximate to a monogenic disorder. Such cases are seen for 9p34 (Kleefstra syndrome) and NRXN1-α, and a case can be made for SHANK3 where individual gene mutations share the same clinical phenotype as the patient with 22q13.2 CNV. However, for most CNVs multiple candidate genes lie within the affected chromosome locus. To progress these cases, we need to establish the mechanism of disease on an individual CNV basis. In principle, this can be achieved by gene transcriptional analysis of the genes within the CNV and cross-reference to GWAS data across the range of NDDs and associated comorbidities. In practice, this can be challenging as multiple genes can often be expressed with patterns that vary from one cell type to another or developmental state; comparison of deletions and duplications at the same loci often do not show “mirror”, or reciprocal expression patterns and GWAS data for NDD is still not saturated for all possible loci.
A further confound is the suspicion that many large CNV clinical phenotypes may arise due to the effects of multiple genes within the locus. Here, patient iPSCs studies may help, as CRISPR-generated isogenic human iPSCs may be directly compared to patient-derived iPSCs. Current genome editing methodologies are already available to allow with the generation of multiple gene manipulations to model these possibilities [112].
Such comparisons can also address patient genetic complexity due to the presence of either multiple CNVs or possession of common variants that elevate the polygenic risk in the genome background. The key to these approaches however will be to have a complete analysis of genomes of patients harbouring pathogenic CNVs, especially if these can be combined with family studies. The most complex genetic scenario is if the structural variation at a CNV alters gene regulation via long-range changes in nuclear architecture or trans-acting regulatory RNA molecules. Again, this would synergise well with patient iPSC approaches via multi-omic methodologies to combine cell phenotyping with genome-wide transcriptional profiling, including miRNAs and lncRNAs, and chromatin interaction studies. Given that GWAS studies indicate that for NDDs 80% of disease-associated SNPs are not within protein-coding genes, such patient iPSC studies may prove a powerful stepping-stone to understanding the impact of the majority of SNPs on inherited ASD risk.
Assay complexity
Although progress has been made, there is still much further work required to improve construct validity of iPSC-based NDD assays. Many neuronal and glial cell types can be generated using available in vitro differentiation protocols. However, cell type diversity is still missing, particularly for the GABAergic interneuron populations, such as the rapid firing PV+ interneurons, which play an important regulatory function and exhibit abnormal behaviour in NDD. Expression of certain combinations of transcription factors can rapidly induce homogeneous populations of glutamatergic, excitatory neurons in the case of NGN2, and GABAergic, inhibitory neurons by expression of Ascl1 and Dlx2 [113]. The expectation is that more transcription factor combinations will be established to enable a greater range of cell type to be generated. However, this approach is not without issues. Given the evidence that the neuronal developmental programme may be disrupted in ASD patient cells, accelerating their neural cell differentiation may bypass some aspects of the cell phenotype. Further work remains to compare iPSC-derived neurons using classical and induced methods to ensure that they accurately reflect those found in the human brain.
In addition to increased assay complexity through generation and co-culture of multiple neuronal and glial cell types, a second element is to create more structured cultures beyond simple growth and development on 2D surfaces. Any increase in cell culture complexity needs to be standardised and deliver robust readouts. The development of 3D brain organoids aims to address this need; however, individual organoids vary considerably in structure and standardised methods of comparison still need to be developed and adopted. A compromise position that has a high degree of controllability, and hence potential for standardisation, is to use 2D cultured cells presented in a layered configuration. Simple examples of this are “sandwich” cultures where neurons and astrocytes are grown on coverslips and dishes as 2D monolayers and then placed together [114]. More sophisticated structured culturing methods are also being developed using 3D bioprinting [115] to build layered cultures where different cells types are set in layers of matrix creating flat interfaces between different cell types.
Finally, complexity can be increased via high content, multiparametric data collection. This can be achieved through increasing the number of parameters recorded for individual assay modalities, such as cellomic approaches with automated cell microscopy, or collection of MEA functional data, all of which are compatible with multi-well formats. Alternatively, it is possible to combine modalities together, so that electrophysiological techniques can be combined with imaging of cell and neuronal gene expression profiles in iPSCs to pinpoint exactly how patient cells differ from non-patient controls. This also requires creation of comparative databases that bring together morphological, genetic, gene expression, biophysical and electrophysiological properties across the neuro-differentiation time course. An exemplar is the Neuroelectro project that aims to compile and organise published data about electrical properties of neurons [116]. This is currently oriented to improve neuronal classification for animal models but could form the basis for a human iPSC platform.
Size and scale
The third dimension is to expand the number of patient iPSCs within experiments, increasing the size of studies from less than 10 patients to many 100s if not 1000s of patients. New automation technologies, such as a fully automated robotic cell reprogramming system [117], are needed to allow the generation of large cohorts of patient-specific iPSCs. These need to be differentiated in parallel with robust and standardised protocols, which would include internal monitoring of the differentiation state.
Many of these issues can be solved by developing higher throughput and analysis culture techniques; however, the greatest limitation is the identification of sufficient numbers of CNV patients. The need to establish large patient cohorts and share data from these rare individuals in the population will be a strong driver for future international collaboration. Research networks, such as the MINDDS COST Action (CA16210) [118], are currently aiming to open up access to larger scale patient-based studies, including those based on iPSCs. As this endeavour grows, there will be the necessity to analyse, collate and share data across multiple sites. The solutions to these problems are already being developed, and projects such as RD-connect [119] may produce the platforms for effective data and resource sharing.
Conclusions
Here, we have considered the need, current state and future of the utility of iPSC-based models of ASD derived from patients with associated CNVs. We have demonstrated that current evidence is accumulating for construct validity between the biological processes that can be studied at the cellular level and clinical observations on patients. We have also discussed the necessity to probe background genetics for iPSC studies of CNV patients, and the opportunity that their cell phenotyping offers for future research to resolve the biological effects of common variants on increased disease risk. Finally, we have considered the future of CNV patient-based cell platforms expanded into the three domains of increased genomic complexity, cell assay complexity and patient population size. While challenging, the expansion of CNV-focused iPSC investigations is feasible and the need to create larger patient iPSC panels will be a driver for future research. This global enterprise will create a unique cell interface to connect ASD risk genetics and clinical phenotype that accelerates the development of personalised medicine.
Availability of data and materials
Not applicable
Abbreviations
- NDD:
-
Neurodevelopmental disorders
- ASD:
-
Autism spectrum disorders
- ID:
-
Intellectual disability
- iPSCs:
-
Induced pluripotent stem cells
- SNVs:
-
Single nucleotide variants
- CNVs:
-
Copy number variants
- WBS:
-
Williams-Beuren syndrome
- PMDS:
-
Phelan-McDermid syndrome
- miRNA:
-
MicroRNA
- lncRNA:
-
Long non-coding RNA
- ADHD:
-
Attention-deficit/hyperactivity disorders
- MEAs:
-
Multi-electrode arrays
References
Zandt F, Prior M, Kyrios M. Repetitive behaviour in children with high functioning autism and obsessive compulsive disorder. J Autism Dev Disord. 2007;37(2):251–9.
Fakhoury M. Autistic spectrum disorders: a review of clinical features, theories and diagnosis. Int J Dev Neurosci. 2015;43:70–7.
Piao Y, Hung SS, Lim SY, Wong RC, Ko MS. Efficient generation of integration-free human induced pluripotent stem cells from keratinocytes by simple transfection of episomal vectors. Stem Cells Transl Med. 2014;3(7):787–91.
Brown ME, Rondon E, Rajesh D, Mack A, Lewis R, Feng X, et al. Derivation of induced pluripotent stem cells from human peripheral blood T lymphocytes. PLoS One. 2010;5(6):e11373.
Seki T, Yuasa S, Fukuda K. Derivation of induced pluripotent stem cells from human peripheral circulating T cells. Curr Protoc Stem Cell Biol. 2011;Chapter 4:Unit4A 3.
Zhou T, Benda C, Dunzinger S, Huang Y, Ho JC, Yang J, et al. Generation of human induced pluripotent stem cells from urine samples. Nat Protoc. 2012;7(12):2080–9.
Zhou T, Benda C, Duzinger S, Huang Y, Li X, Li Y, et al. Generation of induced pluripotent stem cells from urine. J Am Soc Nephrol. 2011;22(7):1221–8.
Velasco S, Kedaigle AJ, Simmons SK, Nash A, Rocha M, Quadrato G, et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature. 2019;570(7762):523–7.
Yoon SJ, Elahi LS, Pasca AM, Marton RM, Gordon A, Revah O, et al. Reliability of human cortical organoid generation. Nat Methods. 2019;16(1):75–8.
Iefremova V, Manikakis G, Krefft O, Jabali A, Weynans K, Wilkens R, et al. An organoid-based model of cortical development identifies non-cell-autonomous defects in Wnt signaling contributing to miller-Dieker syndrome. Cell Rep. 2017;19(1):50–9.
Mellios N, Feldman DA, Sheridan SD, Ip JPK, Kwok S, Amoah SK, et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol Psychiatry. 2018;23(4):1051–65.
Wang P, Mokhtari R, Pedrosa E, Kirschenbaum M, Bayrak C, Zheng D, et al. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol Autism. 2017;8:11.
Lopez-Tobon A, Villa CE, Cheroni C, Trattaro S, Caporale N, Conforti P, et al. Human cortical organoids expose a differential function of GSK3 on cortical neurogenesis. Stem Cell Reports. 2019;13(5):847–61.
Munnich A, Demily C, Frugere L, Duwime C, Malan V, Barcia G, et al. Impact of on-site clinical genetics consultations on diagnostic rate in children and young adults with autism spectrum disorder. Mol Autism. 2019;10:33.
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–9.
Kirov G, Rees E, Walters J. What a psychiatrist needs to know about copy number variants. B J Psych Advances. 2015;21(3):157–63.
Rees E, Walters JT, Georgieva L, Isles AR, Chambert KD, Richards AL, et al. Analysis of copy number variations at 15 schizophrenia-associated loci. Br J Psychiatry. 2014;204(2):108–14.
Kendall KM, Rees E, Escott-Price V, Einon M, Thomas R, Hewitt J, et al. Cognitive performance among carriers of pathogenic copy number variants: analysis of 152,000 UK biobank subjects. Biol Psychiatry. 2017;82(2):103–10.
Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94(5):677–94.
Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148(6):1223–41.
van der Zwaag B, Staal WG, Hochstenbach R, Poot M, Spierenburg HA, de Jonge MV, et al. A co-segregating microduplication of chromosome 15q11.2 pinpoints two risk genes for autism spectrum disorder. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(4):960–6.
Depienne C, Moreno-De-Luca D, Heron D, Bouteiller D, Gennetier A, Delorme R, et al. Screening for genomic rearrangements and methylation abnormalities of the 15q11-q13 region in autism spectrum disorders. Biol Psychiatry. 2009;66(4):349–59.
Serur Y, Sofrin Frumer D, Daon K, Sobol-Havia D, Weinberger R, Shulman C, et al. Psychiatric disorders and autism in young children with 22q11.2 deletion syndrome compared to children with idiopathic autism. Eur Psychiatry. 2019;55:116–21.
Morrow BE, McDonald-McGinn DM, Emanuel BS, Vermeesch JR, Scambler PJ. Molecular genetics of 22q11.2 deletion syndrome. Am J Med Genet A. 2018;176(10):2070–81.
Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch Gen Psychiatry. 1999;56(10):940–5.
Schneider M, Debbane M, Bassett AS, Chow EW, Fung WL, van den Bree M, et al. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the international consortium on brain and behavior in 22q11.2 deletion syndrome. Am J Psychiatry. 2014;171(6):627–39.
Demaerel W, Mostovoy Y, Yilmaz F, Vervoort L, Pastor S, Hestand MS, et al. The 22q11 low copy repeats are characterized by unprecedented size and structural variability. Genome Res. 2019;29(9):1389–401.
Zhang X, Zhang Y, Zhu X, Purmann C, Haney MS, Ward T, et al. Local and global chromatin interactions are altered by large genomic deletions associated with human brain development. Nat Commun. 2018;9(1):5356.
Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med. 2012;367(14):1321–31.
Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42(3):203–9.
Bergen SE, Ploner A, Howrigan D, Group CNVA. The schizophrenia working Group of the Psychiatric Genomics C, O'Donovan MC, et al. joint contributions of rare copy number variants and common SNPs to risk for schizophrenia. Am J Psychiatry. 2019;176(1):29–35.
Martin J, O'Donovan MC, Thapar A, Langley K, Williams N. The relative contribution of common and rare genetic variants to ADHD. Transl Psychiatry. 2015;5:e506.
Velinov M. Genomic copy number variations in the autism clinic-work in progress. Front Cell Neurosci. 2019;13:57.
Bassett AR. Editing the genome of hiPSC with CRISPR/Cas9: disease models. Mamm Genome. 2017;28(7-8):348–64.
Canver MC, Bauer DE, Dass A, Yien YY, Chung J, Masuda T, et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem. 2014;289(31):21312–24.
Tai DJ, Ragavendran A, Manavalan P, Stortchevoi A, Seabra CM, Erdin S, et al. Engineering microdeletions and microduplications by targeting segmental duplications with CRISPR. Nat Neurosci. 2016;19(3):517–22.
Li X, Burnight ER, Cooney AL, Malani N, Brady T, Sander JD, et al. piggyBac transposase tools for genome engineering. Proc Natl Acad Sci U S A. 2013;110(25):E2279–87.
Adhya D, Swarup V, Nagy R, Shum C, Nowosiad P, Jozwik KM, et al. Atypical neurogenesis and excitatory-inhibitory progenitor generation in induced pluripotent stem cell (iPSC) from autistic individuals. bioRxiv. 2019:349415.
Deneault E, Faheem M, White SH, Rodrigues DC, Sun S, Wei W, et al. CNTN5(-)(/+)or EHMT2(-)(/+)human iPSC-derived neurons from individuals with autism develop hyperactive neuronal networks. eLife. 2019;8.
Lam M, Moslem M, Bryois J, Pronk RJ, Uhlin E, Ellstrom ID, et al. Single cell analysis of autism patient with bi-allelic NRXN1-alpha deletion reveals skewed fate choice in neural progenitors and impaired neuronal functionality. Exp Cell Res. 2019;383(1):111469.
Gridina MM, Matveeva NM, Fishman VS, Menzorov AG, Kizilova HA, Beregovoy NA, et al. Allele-Specific Biased Expression of the CNTN6 Gene in iPS Cell-Derived Neurons from a Patient with Intellectual Disability and 3p26.3 Microduplication Involving the CNTN6 Gene. Mol Neurobiol. 2018;55(8):6533–46.
Piovani G, Lanzi G, Ferraro RM, Masneri S, Barisani C, Savio G, et al. Generation of induced pluripotent stem cells (iPSCs) from patient with Cri du Chat Syndrome. Stem Cell Res. 2019;35:101393.
Adamo A, Atashpaz S, Germain PL, Zanella M, D'Agostino G, Albertin V, et al. 7q11.23 dosage-dependent dysregulation in human pluripotent stem cells affects transcriptional programs in disease-relevant lineages. Nat Genet. 2015;47(2):132–41.
Frega M, Linda K, Keller JM, Gumus-Akay G, Mossink B, van Rhijn JR, et al. Neuronal network dysfunction in a model for Kleefstra syndrome mediated by enhanced NMDAR signaling. Nat Commun. 2019;10(1):4928.
Willemsen MH, Beunders G, Callaghan M, de Leeuw N, Nillesen WM, Yntema HG, et al. Familial Kleefstra syndrome due to maternal somatic mosaicism for interstitial 9q34.3 microdeletions. Clin Genet. 2011;80(1):31–8.
Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell. 2015;162(2):375–90.
Abyzov A, Mariani J, Palejev D, Zhang Y, Haney MS, Tomasini L, et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature. 2012;492(7429):438–42.
Das DK, Tapias V, D'Aiuto L, Chowdari KV, Francis L, Zhi Y, et al. Genetic and morphological features of human iPSC-derived neurons with chromosome 15q11.2 (BP1-BP2) deletions. Mol Neuropsychiatry. 2015;1(2):116–23.
Arioka Y, Kushima I, Mori D, Ozaki N. Three lines of induced pluripotent stem cells derived from a 15q11.2-q13.1 duplication syndrome patient. Stem Cell Res. 2018;31:240–3.
Germain ND, Chen PF, Plocik AM, Glatt-Deeley H, Brown J, Fink JJ, et al. Gene expression analysis of human induced pluripotent stem cell-derived neurons carrying copy number variants of chromosome 15q11-q13.1. Mol Autism. 2014;5:44.
Gillentine MA, Yin J, Bajic A, Zhang P, Cummock S, Kim JJ, et al. Functional consequences of CHRNA7 copy-number alterations in induced pluripotent stem cells and neural progenitor cells. Am J Hum Genet. 2017;101(6):874–87.
Deshpande A, Yadav S, Dao DQ, Wu ZY, Hokanson KC, Cahill MK, et al. Cellular phenotypes in human iPSC-derived neurons from a genetic model of autism spectrum disorder. Cell Rep. 2017;21(10):2678–87.
Johnstone M, Vasistha NA, Barbu MC, Dando O, Burr K, Christopher E, et al. Reversal of proliferation deficits caused by chromosome 16p13.11 microduplication through targeting NFkappaB signaling: an integrated study of patient-derived neuronal precursor cells, cerebral organoids and in vivo brain imaging. Mol Psychiatry. 2019;24(2):294–311.
Bershteyn M, Nowakowski TJ, Pollen AA, Di Lullo E, Nene A, Wynshaw-Boris A, et al. Human iPSC-derived cerebral organoids model cellular features of lissencephaly and reveal prolonged mitosis of outer radial glia. Cell Stem Cell. 2017;20(4):435–49 e4.
Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503(7475):267–71.
Kathuria A, Nowosiad P, Jagasia R, Aigner S, Taylor RD, Andreae LC, et al. Stem cell-derived neurons from autistic individuals with SHANK3 mutation show morphogenetic abnormalities during early development. Mol Psychiatry. 2018;23(3):735–46.
Cocks G, Curran S, Gami P, Uwanogho D, Jeffries AR, Kathuria A, et al. The utility of patient specific induced pluripotent stem cells for the modelling of Autistic Spectrum Disorders. Psychopharmacology. 2014;231(6):1079–88.
Ross PJ, Zhang WB, Mok RSF, Zaslavsky K, Deneault E, D'Abate L, et al. Synaptic dysfunction in human neurons with autism-associated deletions in PTCHD1-AS. Biol Psychiatry. 2019.
Machado CO, Griesi-Oliveira K, Rosenberg C, Kok F, Martins S, Passos-Bueno MR, et al. Collybistin binds and inhibits mTORC1 signaling: a potential novel mechanism contributing to intellectual disability and autism. Eur J Hum Genet. 2016;24(1):59–65.
Nageshappa S, Carromeu C, Trujillo CA, Mesci P, Espuny-Camacho I, Pasciuto E, et al. Altered neuronal network and rescue in a human MECP2 duplication model. Mol Psychiatry. 2016;21(2):178–88.
Buckholtz JW, Meyer-Lindenberg A. Psychopathology and the human connectome: toward a transdiagnostic model of risk for mental illness. Neuron. 2012;74(6):990–1004.
Falk A, Heine VM, Harwood AJ, Sullivan PF, Peitz M, Brustle O, et al. Modeling psychiatric disorders: from genomic findings to cellular phenotypes. Mol Psychiatry. 2016;21(9):1167–79.
Gouder L, Vitrac A, Goubran-Botros H, Danckaert A, Tinevez JY, Andre-Leroux G, et al. Altered spinogenesis in iPSC-derived cortical neurons from patients with autism carrying de novo SHANK3 mutations. Sci Rep. 2019;9(1):94.
DeRosa BA, El Hokayem J, Artimovich E, Garcia-Serje C, Phillips AW, Van Booven D, et al. Convergent pathways in idiopathic autism revealed by time course transcriptomic analysis of patient-derived neurons. Sci Rep. 2018;8(1):8423.
Schafer ST, Paquola ACM, Stern S, Gosselin D, Ku M, Pena M, et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat Neurosci. 2019;22(2):243–55.
Mozzetta C, Pontis J, Fritsch L, Robin P, Portoso M, Proux C, et al. The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol Cell. 2014;53(2):277–89.
Khattak S, Brimble E, Zhang W, Zaslavsky K, Strong E, Ross PJ, et al. Human induced pluripotent stem cell derived neurons as a model for Williams-Beuren syndrome. Mol Brain. 2015;8(1):77.
Huang G, Chen S, Chen X, Zheng J, Xu Z, Doostparast Torshizi A, et al. Uncovering the functional link between SHANK3 deletions and deficiency in neurodevelopment ising iPSC-derived human neurons. Front Neuroanat. 2019;13:23.
Siniscalco D, Schultz S, Brigida AL, Antonucci N. Inflammation and neuro-immune dysregulations in autism spectrum disorders. Pharmaceuticals. 2018;11(2).
Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. 2014;17(3):400–6.
Haan N, Carter J, Westacott LJ, Owen MJ, Gray WP, Hall J, et al. Haploinsufficiency of the schizophrenia risk gene Cyfip1 causes abnormal postnatal hippocampal neurogenesis through a novel microglia dependent mechanism. bioRxiv. 2018:417832.
Russo FB, Freitas BC, Pignatari GC, Fernandes IR, Sebat J, Muotri AR, et al. Modeling the interplay between neurons and astrocytes in autism using human induced pluripotent stem cells. Biol Psychiatry. 2018;83(7):569–78.
Eftekharian MM, Ghafouri-Fard S, Noroozi R, Omrani MD, Arsang-Jang S, Ganji M, et al. Cytokine profile in autistic patients. Cytokine. 2018;108:120–6.
Nadadhur AG, Alsaqati M, Gasparotto L, Cornelissen-Steijger P, van Hugte E, Dooves S, et al. Neuron-glia interactions increase neuronal phenotypes in tuberous sclerosis complex patient iPSC-derived models. Stem Cell Reports. 2019;12(1):42–56.
Courchesne E, Carper R, Akshoomoff N. Evidence of brain overgrowth in the first year of life in autism. Jama. 2003;290(3):337–44.
Courchesne E, Karns CM, Davis HR, Ziccardi R, Carper RA, Tigue ZD, et al. Unusual brain growth patterns in early life in patients with autistic disorder: an MRI study. Neurology. 2001;57(2):245–54.
Lainhart JE, Piven J, Wzorek M, Landa R, Santangelo SL, Coon H, et al. Macrocephaly in children and adults with autism. J Am Acad Child Adolesc Psychiatry. 1997;36(2):282–90.
Piven J, Arndt S, Bailey J, Havercamp S, Andreasen NC, Palmer P. An MRI study of brain size in autism. Am J Psychiatry. 1995;152(8):1145–9.
Lin A, Ching CRK, Vajdi A, Sun D, Jonas RK, Jalbrzikowski M, et al. Mapping 22q11.2 gene dosage effects on brain morphometry. J Neurosci. 2017;37(26):6183–99.
Sun D, CRK C, Lin A, Forsyth JK, Kushan L, Vajdi A, et al. Large-scale mapping of cortical alterations in 22q11.2 deletion syndrome: convergence with idiopathic psychosis and effects of deletion size. Mol Psychiatry. 2018.
Fan CC, Brown TT, Bartsch H, Kuperman JM, Hagler DJ Jr, Schork A, et al. Williams syndrome-specific neuroanatomical profile and its associations with behavioral features. NeuroImage Clin. 2017;15:343–7.
Meda SA, Pryweller JR, Thornton-Wells TA. Regional brain differences in cortical thickness, surface area and subcortical volume in individuals with Williams syndrome. PLoS One. 2012;7(2):e31913.
Silva AI, Ulfarsson MO, Stefansson H, Gustafsson O, Walters GB, Linden DEJ, et al. Reciprocal white matter changes associated with copy number variation at 15q11.2 BP1-BP2: a diffusion tensor imaging study. Biol Psychiatry. 2019;85(7):563–72.
Ulfarsson MO, Walters GB, Gustafsson O, Steinberg S, Silva A, Doyle OM, et al. 15q11.2 CNV affects cognitive, structural and functional correlates of dyslexia and dyscalculia. Transl Psychiatry. 2017;7(4):e1109.
Writing Committee for the E-CNVWG, van der Meer D, Sonderby IE, Kaufmann T, Walters GB, Abdellaoui A, et al. Association of copy number variation of the 15q11.2 BP1-BP2 region with cortical and subcortical morphology and cognition. JAMA Psychiatry. 2019;77:1–11.
Maillard AM, Ruef A, Pizzagalli F, Migliavacca E, Hippolyte L, Adaszewski S, et al. The 16p11.2 locus modulates brain structures common to autism, schizophrenia and obesity. Mol Psychiatry. 2015;20(1):140–7.
Martin-Brevet S, Rodriguez-Herreros B, Nielsen JA, Moreau C, Modenato C, Maillard AM, et al. Quantifying the effects of 16p11.2 copy number variants on brain structure: a multisite genetic-first study. Biol Psychiatry. 2018;84(4):253–64.
Qureshi AY, Mueller S, Snyder AZ, Mukherjee P, Berman JI, Roberts TP, et al. Opposing brain differences in 16p11.2 deletion and duplication carriers. J Neurosci. 2014;34(34):11199–211.
Sonderby IE, Gustafsson O, Doan NT, Hibar DP, Martin-Brevet S, Abdellaoui A, et al. Dose response of the 16p11.2 distal copy number variant on intracranial volume and basal ganglia. Mol Psychiatry. 2020;25(3):584–602.
Chen CP, Keown CL, Jahedi A, Nair A, Pflieger ME, Bailey BA, et al. Diagnostic classification of intrinsic functional connectivity highlights somatosensory, default mode, and visual regions in autism. Neuroimage Clin. 2015;8:238–45.
Haar S, Berman S, Behrmann M, Dinstein I. Anatomical abnormalities in autism? Cereb Cortex. 2016;26(4):1440–52.
Marchetto MC, Belinson H, Tian Y, Freitas BC, Fu C, Vadodaria K, et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry. 2017;22(6):820–35.
Coghlan S, Horder J, Inkster B, Mendez MA, Murphy DG, Nutt DJ. GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neurosci Biobehav Rev. 2012;36(9):2044–55.
Ecker C, Suckling J, Deoni SC, Lombardo MV, Bullmore ET, Baron-Cohen S, et al. Brain anatomy and its relationship to behavior in adults with autism spectrum disorder: a multicenter magnetic resonance imaging study. Arch Gen Psychiatry. 2012;69(2):195–209.
Lenroot RK, Yeung PK. Heterogeneity within autism spectrum disorders: what have we learned from neuroimaging studies? Front Hum Neurosci. 2013;7:733.
Zikopoulos B, Barbas H. Changes in prefrontal axons may disrupt the network in autism. J Neurosci. 2010;30(44):14595–609.
Nuninga JO, Bohlken MM, Koops S, Fiksinski AM, Mandl RCW, Breetvelt EJ, et al. White matter abnormalities in 22q11.2 deletion syndrome patients showing cognitive decline. Psychol Med. 2018;48(10):1655–63.
Drakesmith M, Parker GD, Smith J, Linden SC, Rees E, Williams N, et al. Genetic risk for schizophrenia and developmental delay is associated with shape and microstructure of midline white-matter structures. Transl Psychiatry. 2019;9(1):102.
Silva AI, Haddon JE, Ahmed Syed Y, Trent S, Lin TE, Patel Y, et al. Cyfip1 haploinsufficient rats show white matter changes, myelin thinning, abnormal oligodendrocytes and behavioural inflexibility. Nat Commun. 2019;10(1):3455.
Hussein SM, Batada NN, Vuoristo S, Ching RW, Autio R, Narva E, et al. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011;471(7336):58–62.
Draper JS, Smith K, Gokhale P, Moore HD, Maltby E, Johnson J, et al. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat Biotechnol. 2004;22(1):53–4.
International Stem Cell I, Amps K, Andrews PW, Anyfantis G, Armstrong L, Avery S, et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat Biotechnol. 2011;29(12):1132–44.
Laurent LC, Ulitsky I, Slavin I, Tran H, Schork A, Morey R, et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell. 2011;8(1):106–18.
Maitra A, Arking DE, Shivapurkar N, Ikeda M, Stastny V, Kassauei K, et al. Genomic alterations in cultured human embryonic stem cells. Nat Genet. 2005;37(10):1099–103.
Mertens J, Paquola ACM, Ku M, Hatch E, Bohnke L, Ladjevardi S, et al. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell. 2015;17(6):705–18.
Sheng C, Jungverdorben J, Wiethoff H, Lin Q, Flitsch LJ, Eckert D, et al. A stably self-renewing adult blood-derived induced neural stem cell exhibiting patternability and epigenetic rejuvenation. Nat Commun. 2018;9(1):4047.
Prigione A, Hossini AM, Lichtner B, Serin A, Fauler B, Megges M, et al. Mitochondrial-associated cell death mechanisms are reset to an embryonic-like state in aged donor-derived iPS cells harboring chromosomal aberrations. PLoS One. 2011;6(11):e27352.
Perrera V, Martello G. How does reprogramming to pluripotency affect genomic imprinting? Front Cell Dev Biol. 2019;7:76.
Xie Y, Schutte RJ, Ng NN, Ess KC, Schwartz PH, O'Dowd DK. Reproducible and efficient generation of functionally active neurons from human hiPSCs for preclinical disease modeling. Stem Cell Res. 2018;26:84–94.
van den Hurk M, Erwin JA, Yeo GW, Gage FH, Bardy C. Patch-Seq protocol to analyze the electrophysiology, morphology and transcriptome of whole single neurons derived from human pluripotent stem cells. Front Mol Neurosci. 2018;11:261.
Trujillo CA, Gao R, Negraes PD, Gu J, Buchanan J, Preissl S, et al. Complex oscillatory waves emerging from cortical organoids model early human brain network development. Cell Stem Cell. 2019;25(4):558–69 e7.
Schrode N, Ho SM, Yamamuro K, Dobbyn A, Huckins L, Matos MR, et al. Synergistic effects of common schizophrenia risk variants. Nat Genet. 2019;51(10):1475–85.
Yang N, Chanda S, Marro S, Ng YH, Janas JA, Haag D, et al. Generation of pure GABAergic neurons by transcription factor programming. Nat Methods. 2017;14(6):621–8.
Boraso M, Viviani B. Glia-neuron sandwich cocultures: an in vitro approach to evaluate cell-to-cell communication in neuroinflammation and neurotoxicity. Methods Mol Biol. 2011;758:135–52.
Salaris F, Colosi C, Brighi C, Soloperto A, Turris V, Benedetti MC, et al. 3D bioprinted human cortical neural constructs derived from induced pluripotent stem cells. J Clin Med. 2019;8(10):1595.
Tripathy SJ, Savitskaya J, Burton SD, Urban NN, Gerkin RC. NeuroElectro: a window to the world's neuron electrophysiology data. Front Neuroinformatics. 2014;8:40.
Paull D, Sevilla A, Zhou H, Hahn AK, Kim H, Napolitano C, et al. Automated, high-throughput derivation, characterization and differentiation of induced pluripotent stem cells. Nat Methods. 2015;12(9):885–92.
European COST Action CA16210 Maximizing Impact of research of NeuroDevelopmental DisorderS (MINDDS). Available from: https://mindds.eu/.
Thompson R, Johnston L, Taruscio D, Monaco L, Beroud C, Gut IG, et al. RD-connect: an integrated platform connecting databases, registries, biobanks and clinical bioinformatics for rare disease research. J Gen Intern Med. 2014;29(Suppl 3):S780–7.
Acknowledgements
The authors wish to acknowledge the support of the European COST Action CA16210 MINDDS for a Workshop to organize the writing of this review. In addition, the authors would like to thank Dr. Ida E. Sønderby for participating in the preparation of the manuscript.
Funding
DD was supported by the Ministry of Education, Science and Technological Development, Republic of Serbia (grant no. 173051 and agreement no. 451-03-68/2020-14/200042). SD was supported by the South-Eastern Norway Regional Health Authority (no. 2018094). ARM was funded by the Spanish “Agencia Estatal de Investigación” (BFU2015-68655-P) and AF by SSF (IB13-0074), VR (2017-03407), Hjärnfonden. SP was supported by the Hellenic Foundation for Research and Innovation (HFRI) and the General Secretariat for Research and Technology (GSRT) [grant agreement no. 122]. All authors were supported by European COST Action, CA16210 Maximizing Impact of research in NeuroDevelopmental DisorderS (MINDDS).
Author information
Authors and Affiliations
Contributions
DD, SD, YAS, ST, NC, AF, RO, VMH, SM, ARM, MB, GT, SJRAC, SP, and AJH collected information and wrote the manuscript. DD, SP, MBMvdB, and AJH edited the manuscript. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplementary Table 2a.
iPSCs generated from patients carrying CNVs and were diagnosed with ASD.
Additional file 2: Supplementary Table 2b.
iPSCs generated from patients not diagnosed with ASD but carrying CNVs which might be associated with NDD.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Drakulic, D., Djurovic, S., Syed, Y.A. et al. Copy number variants (CNVs): a powerful tool for iPSC-based modelling of ASD. Molecular Autism 11, 42 (2020). https://doi.org/10.1186/s13229-020-00343-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13229-020-00343-4