
Abstract
Background
Reactive oxygen species may be involved in epigenetic gene activation or silencing. We aimed to identify CpG sites, at which DNA methylation is related to urinary 8-isoprostane levels (biomarker of lipid peroxidation) and cancer or mortality outcomes. This investigation was based on a German, population-based cohort with linkage to cancer and mortality registry data (2000–2016).
Results
Blood DNA methylation in promoter regions of 519 genes, known to be involved in pathways from oxidative stress (OS) to cancer, was obtained at the cohort's baseline examination. Inverse associations of DNA methylation at cg25365794 (ALOXE3) and cg08862778 (MTOR) with 8-isoprostane levels were observed in a derivation set (n = 1000) and validated in two independent subsets of the cohort (n = 548 and n = 741). Multivariate regression models were used to evaluate the associations of DNA methylation at the two CpG sites with lung, colorectal, prostate, breast, and overall cancer incidence as well as CVD, cancer, and all-cause mortality. DNA methylation at cg25365794 (ALOXE3) was inversely associated with lung and prostate cancer incidence. DNA methylation at cg08862778 (MTOR) was associated with a 43% lower breast cancer incidence in the top vs. bottom tertile.
Conclusion
The finding for ALOXE3 may not be causal. As ALOXE3 is mainly expressed in skin tissue, the observed association might reflect the fact that both DNA methylation at the ALOXE3 gene and urinary 8-isoprostane concentrations depend on the level of OS in tissues. Contrarily, the finding for the MTOR gene and breast cancer is biologically plausible because the MTOR protein plays an important role in PI3K/Akt signaling, which is a pathway related to cancer development and cell senescence.
Similar content being viewed by others


Background
Oxidative stress (OS) refers to an imbalance between antioxidants and oxidants production in favor of the latter, resulting in an interruption of redox signaling and control and/or molecular damage [1]. OS at physiological levels is termed oxidative eustress, and maintenance of oxidative eustress is crucial for redox regulation [2, 3]. Excessive OS, however, can damage proteins, lipids, and nucleic acids, can contribute to the development of various age-related diseases, including cancer [4]. The major oxidants are reactive oxygen species (ROS), which cannot be detected directly in human specimens because of their short half-lives [5]. Instead, elevated ROS can be indirectly measured by using oxidatively generated metabolites of proteins, lipids, or nucleic acids. The concentration of 8-isoprostane molecules in urine, a biomarker of lipid peroxidation, is the acknowledged gold standard for measurement of the oxidative stress burden of the human organism [6, 7]. Elevated 8-isoprostane levels have been shown to be a risk factor for lung cancer [8, 9].
Excessive ROS has been shown to be involved in the changes of DNA methylation levels [10, 11]. For instance, hydrogen peroxide acts as a nucleophile to deprotonate the cytosine molecule at the C-5 position, which accelerates the reaction of DNA with the positive-charged intermediate S-adenosyl-L-methionine in the process of DNA methylation [12]. In addition, ROS may regulate the expression of DNA methyltransferases (DNMT), enzymes catalyzing the transfer of a methyl group to DNA [13]. Conversely, DNA methylation alterations may regulate the expression of OS-related genes [14]. In spite of biological plausibility, evidence from human population-based studies on the associations of OS biomarker concentrations with DNA methylation of OS-related genes is sparse and the potential impact of OS-related DNA methylation alteration on cancer development is even sparser [15, 16].
Therefore, this study aimed (i) to conduct a gene-specific screening for CpG sites, differentially methylated according to urinary 8-isoprostane levels, and (ii) to investigate whether identified CpG sites are associated with the risk of cancer or mortality.
Methods
Study population
This study is based on the ESTHER study (German: Epidemiologische Studie zu Chancen der Verhütung, Früherkennung und optimierten Therapie chronischer Erkrankungen in der älteren Bevölkerung), which is an ongoing prospective, population-based cohort study. Details of the study design have been reported elsewhere [17, 18]. Briefly, general practitioners, (GPs) recruited 9949 study participants aged 50 to 74 years, during a general health check-up between 2000 and 2002 in Saarland, a federal state of Germany. Spot urine and whole blood samples were collected during the health check-up, shipped to the study center, and maintained at − 80 °C until further processing.
Four independent subsets were selected from the ESTHER study for epigenome-wide DNA methylation data measurements for various projects [19] and were used for the current analysis (Fig. 1). Subset I includes the first 500 recruited men and the first 500 recruited women (recruited between July and October 2000). Subset II has a case-cohort design for mortality with n = 316 deaths and a random sub-cohort of n = 548. Subset III has a case-cohort design with n = 128 breast cancer, n = 58 lung cancer, n = 23 colorectal cancer, and n = 538 mortality cases and a random sub-cohort with n = 741 subjects. Subset IV has a nested case-control design with n = 65 incident lung cancer cases, n = 100 colorectal cancer cases, and n = 176 controls. In the gene-specific screening for CpG sites, differentially methylated according to urinary 8-isoprostane levels, subset I was used as the derivation set and the random sub-cohorts of subset II and III were used as two independent validation sets. Finally, all four subsets were used to determine potential associations of identified CpG sites with cancer (lung, colorectal, breast, prostate, and overall cancer) and mortality outcomes (cancer, CVD, and all-cause mortality).

Four subsets of the German ESTHER study selected for measurement of epigenome-wide DNA methylation data, Abbreviation: BC, breast cancer; CRC, colorectal cancer; LC, lung cancer
Laboratory analyses
Urinary 8-isoprostane levels were determined by 8iso1 ELISA kit from Detroit R&D (Detroit, Michigan, USA) with two- or fourfold dilution, depending on the concentrations of the marker. According to the manufacturer’s manual, the specificity of the assay is 100%. For renal function adjustment of spot urine samples, urinary creatinine was determined by the kinetic Jaffe method and 8-isoprostane levels were expressed with the unit “nmol/mmol creatinine.”
DNA methylation profiles of subset I, II, and IV were assessed with the Infinium HumanMethylation450 BeadChip (450 k) array, and DNA methylation profiles of subset III were assessed with the Infinium Methylation EPIC BeadChip kit that covers 850,000 CpG sites (850k) (Illumina, San Diego, CA, USA). The assays were conducted by following the manufacturer’s instruction at the Genomics and Proteomics Core Facility of the German Cancer Research Center, Heidelberg, Germany [20]. The methylation status of a specific CpG site was quantified as a β-value ranging from 0 (no methylation) to 1 (full methylation). No background correction was done, and data were normalized to internal controls provided by the manufacturer. All controls were checked for inconsistencies in each measured plate. Signals of probes with a detection P value > 0.05 were excluded from analysis.
Outcome ascertainment
Incident cancers until the end of 2014 were ascertained by linkage with the Saarland Cancer Registry. According to the 10th Revision of the International Statistical Classification of Diseases (ICD-10), cancer cases during follow-up were defined by all ICD-10 C-codes but C44 (non-melanoma skin cancer). Colorectal, lung, breast, and prostate cancer were defined by the ICD-10 codes C18-C21, C34, C50, and C61, respectively.
Deaths during follow-up by the end of 2015 were ascertained by inquiry at the residents’ registration offices, and information on the vital status of 99.9% of the cohort's participants could be obtained. Additionally, death certificates were provided by local health authorities for 97.7% of those who had died. All deaths coded with ICD-10 codes I00–I99 were considered cardiovascular deaths, and cancer deaths were defined by ICD-10 codes C00–C99 and D37–D48.
Covariates assessment
Information on sociodemographic characteristics; smoking behavior; physical activity; the consumption of alcohol, fruits, vegetables, and meat; an asthma diagnosis; and history of cardiovascular events (stroke, myocardial infarction, pulmonary embolism, bypass operation, or dilatation of the coronary vessels) were obtained from a standardized self-administered questionnaire. Height, weight, and a history of diabetes or coronary heart disease (CHD) were assessed and documented on a standardized form by GPs during the health check-up. The history of cancer before baseline was determined by either self-report or record linkage with data from the Saarland Cancer Registry, which started to record cancers in 1970.
Selection of CpG site candidates
To increase the statistical power, a gene-specific search was performed with restriction to genes coding for proteins that are involved in intracellular ROS generating organelles and enzymes and signal transduction cascades kinases/phosphatases or transcription factors that are on pathways from increased OS to cancer development. These proteins have been identified by our group in a systematic literature review and reported previously [21]. Altogether, 542 genes involved in 18 pathways were identified. We excluded 8 genes, which were not included in the 450k array, and 15 genes, which were on the X chromosome, leaving 519 genes for analyses. The 3811 CpG sites in the promoter regions of these 519 genes were selected for the screening in the derivation set. The selection process of the CpG sites is illustrated in Fig. 2, and the 519 selected genes and their pathways are listed in Additional file 1: Table S1.

Flow chart of CpG sites selection, Abbreviation: FDR, false discovery rate
Statistical analyses
Baseline characteristics of participants of the derivation sample and the two validation samples were expressed as medians (interquartile ranges) or proportions, and differences among the three samples were determined by Wilcoxon-Mann-Whitney tests for continuous variables and by chi-square tests for discrete variables.
In the derivation set, a mixed linear regression model was used to assess the associations between the methylation levels of the selected 3811 CpG sites and 8-isoprostane levels. A natural logarithm transformation of 8-isoprostane levels was employed to ensure normal distribution. The model was adjusted for age (continuously), sex (male/female), leukocyte composition using Houseman’s algorithm [22] (6 continuous variables), alcohol consumption (continuously), body mass index (BMI, continuously), physical activity (inactive, low, medium, or high), fruit consumption (</≥ once/day), vegetables consumption (</≥ once/day), meat consumption (</≥ once/day), smoking status (with seven categories, as shown in Table 1), history of cancer (yes/no), cardiovascular diseases (yes/no), diabetes (yes/no), and asthma (yes/no). Batch-specific variations of the DNA methylation assay were modeled as random effects.
The top 10 CpG sites with the lowest P values for the association with 8-isoprostane levels in the derivation set were selected, and the same linear mixed regression model analyses were repeated in the two validation sets. Obtained β-coefficients and standard errors were subsequently pooled by fixed effects meta-analysis. To account for multiple testing, only CpG sites with a false discovery rate (FDR) < 0.05 were considered to be statistically significant and selected for analyses with cancer and mortality outcomes.
To explore the associations between DNA methylation at the selected CpG sites and cancer incidences (overall, lung, colorectal, breast, and prostate cancer) as well as mortality outcomes (all-cause, cancer-specific and cardiovascular disease-specific mortality), Cox regression, weighted Cox regression [20], and logistic regression models were used for cohort (subset I and random sub-cohorts of subset II and II), case-cohort (subset II and III), and nested case-control study designs (subset IV), respectively. Hazard ratios (HRs) with corresponding 95% confidence intervals (95% CIs) were estimated in subset I–III, and odds ratios (ORs) with 95% CIs were assessed in the subset IV and results were pooled by fixed effects meta-analysis. Subjects with a history of the specific cancer of interest were excluded. In a sensitivity analysis, also cancer cases that occurred in the first 2 years of follow-up (potentially undiagnosed cancers at baseline) were excluded, but overall, the results did not change (data not shown). The regression models were adjusted for age, sex, leukocyte composition, and batches. Additional potential confounders, to be adjusted for in the regression models, needed to be statistically significantly associated with DNA methylation levels at the selected CpG site (P < 0.05). The potential confounders were identified among all variables shown in Table 1 in general linear models. Methylation intensities were included into the models either as tertiles to test for non-linear associations (bottom tertiles defined as the reference category) or as continuous variables to test for linear associations.
Multiple imputation was applied to adequately deal with missing values, and five data sets were imputed. The variables for the imputation model were those shown in Table 1. No variable had more than 10% missing information. The multiple imputation assumption (values missing at random) was examined, and individuals with complete data did not differ from those with incomplete data (data not shown). All statistical tests were conducted with the Statistical Analysis System (SAS, version 9.4, Cary, NC, USA).
Results
Table 1 presents the baseline characteristics of participants in derivation set and the two validation sets. Subjects were on average 62 years old. Because of different sampling strategies, the derivation set includes equal numbers of men and women and the validation sets have a higher proportion of women than men. The different sex distribution resulted in some statistically significant differences in baseline characteristics (categorized BMI, asthma, 8-isoprostane levels, leukocyte composition), but otherwise, the characteristics of the three samples were comparable.
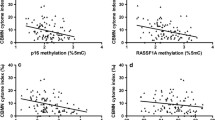
From the 3811 CpG sites in the promoter regions of the 519 selected genes, information on the top 10 CpG sites associated with 8-isoprostane concentrations in the derivation set are shown in Additional file 1: Table S2 (P value ≤ 7.05 × 10−4; FDR ≤ 0.227) and the pooled results from the two validation sets are shown in Table 2. DNA methylation levels at three CpG sites were statistically significantly associated with 8-isoprostane concentrations after FDR correction: cg25365794 [Arachidonate LipOXygEnase 3 (ALOXE3) gene], cg01009697 [Neurotrophic Receptor Tyrosine Kinase 2 (NTRK2) gene], and cg08862778 [Mechanistic target of rapamycin kinase (MTOR) gene]. However, the methylation levels at cg01009697 were positively associated with 8-isoprostane levels in the deviation set (Additional file 1: Table S2) and inversely associated with 8-isoprostane levels in the validation sets (Table 2), which means that the results in the derivation set were not confirmed in the validation sets. Therefore, cg01009697 was excluded from further analyses. The distribution of the methylation levels of the selected two CpG sites in the three subsets is shown in Additional file 1: Figure S1. The linear inverse associations of the methylation levels at the two selected CpG sites with 8-isoprostane levels are additionally shown graphically in a scatter plot in Additional file 1: Figure S2. The explained variance of 8-isoprostane levels by DNA methylation at both CpG sites was rather low (R2 ranged from 0.0015 to 0.0148 in the three subsets). None of the baseline characteristics of the study participants was statistically significantly associated with DNA methylation at the two selected CpG sites (data not shown).
The identified two CpG sites were carried forward to the testing for associations of OS-related DNA methylation with cancer and mortality outcomes (Table 3). Every one standard deviation (SD) increase in DNA methylation at cg25365794 (ALOXE3 gene) resulted in a 19% decrease in incidence of lung cancer (HR (95%) 0.81 (0.66, 0.99)). Furthermore, an inverse association of cg25365794 (ALOXE3 gene) with prostate cancer was observed (HR (95% CI) per 1 SD increase: 0.78 (0.60, 1.03)), but only the comparison of the middle and the bottom tertile was statistically significant (HR (95% CI) 0.47 (0.24, 0.92)). DNA methylation at cg08862778 (MTOR gene) was statistically inversely associated with breast cancer (top tertile vs. bottom tertile, HR (95% CI) 0.57 (0.33, 0.97)).
Discussion
In summary, based on this gene-promoter-specific screening analysis in three independent subsets of the ESTHER cohort, DNA methylation at cg25365794 (ALOXE3 gene) and cg08862778 (MTOR gene) where inversely associated with 8-isoprostane levels. In further analysis, association of DNA methylation at the two selected CpG sites with cancer or mortality outcomes was explored in four subsets has been meta-analyzed. DNA methylation at cg25365794 (ALOXE3 gene) was inversely associated with lung and prostate cancer. Moreover, an inverse association was found between DNA methylation at cg08862778 (MTOR gene) and breast cancer.
The ALOXE3 gene encodes arachidonate lipoxygenase 3, which converts polyunsaturated fatty acid hydroperoxides via an alkoxyl radical intermediate to epoxyalcohols and ketons [23]. These products play an indispensable role in formation of the water-impermeable barrier of the outer epidermis [24]. As ALOXE3 is mainly expressed in skin tissue and to a lesser extent in several other tissues [25], a direct link to 8-isoprostane concentrations in urine is rather unlikely. The observed significant correlation between DNA methylation at the ALOXE3 gene and 8-isoprostane concentrations might reflect the fact that both depend on the level of OS in tissues. Taken together, the observed associations of DNA methylation at the ALOXE3 gene and lung and prostate cancer development might not be causal. However, the currently missing biological plausibility may be found by future studies. To our knowledge, only one population-based study linked ALOXE3 gene and cancer so far and showed that men with mutations in the ALOXE3 gene can have a dysfunctional epidermis barrier and may have an increased risk for prostate cancer if exposed to pesticides [26].
The MTOR gene encodes a serine-threonine protein kinase (mTOR) serving as a core component of two multi-protein complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), which respond to stressors, including DNA damage, nutrients, and oxidative stress [27, 28]. Hydrogen peroxide, a major product of oxidative stress, can activate the PI3K/AKT/mTOR signaling pathway by inhibiting its suppressor, PTEN (phosphatase and tensin homolog) [21]. The PI3K/Akt axis is involved in the regulation of the mTORC1. Activation of PI3K leads to phosphorylation and activation of Akt, which subsequently activates mTORC1 [29]. Over-activation of mTORC1 leads to widespread but benign tumor formation [28]. The mTORC2 protein acts as a tyrosine kinase and phosphorylates Akt, which has a function in maintaining cancer cell survival [30, 31]. Therefore, the PI3K/Akt/mTOR signaling is a target for the treatment of cancer. For instance, everolimus is an mTOR inhibitor and was approved for breast cancer treatment [32]. Supporting our findings, Tang et al. observed that a CpG site located in the regulatory associated protein of MTOR complex 1 (RPTOR) gene, which is an activator of mTOR, was hypomethylated in breast cancer patients compared to healthy controls [33]. Therefore, our finding adds evidence to a pathway of ROS-mediated PI3K/AKT/mTOR activation to breast cancer development. This pathway might be in part controlled by DNA demethylation in the promoter region of the MTOR gene.
To our knowledge, our study is the first gene-specific study, which screened for associations of DNA methylation with 8-isoprostane levels. There is only one other similar study, which used a different biomarker of OS (derivatives of reactive oxygen metabolites (d-ROM)) and had a much smaller sample size (n = 99 in derivation set, n = 142 in validation set) [15]. In this epigenome-wide screening study, DNA methylation at cg10342304 (nucleoredoxin (NXN) gene) was associated with derivatives of reactive oxygen metabolites (d-ROM) and, moreover, significantly associated with overall cancer incidence [15]. A further study that measured DNA methylation in four oxidative stress-related genes identified a significant association of DNA methylation at one site in the promotor region of the 8-oxoguanine DNA glycosylase (OGG1) gene with overall cancer incidence and prostate cancer incidence [16]. However, the results were neither corrected for multiple testing nor validated in another sample. The choice of different OS biomarkers in these two studies may explain why we did not observe associations with DNA methylation at the NXN and the OGG1 gene.
Our analysis has a number of strengths. First, detailed information on a broad range of covariates enabled us to control for potential confounding as far as possible. Second, its prospective design for the cancer outcomes precluded reverse causality. Third, usage of almost complete cancer registry and mortality follow-up data excluded misclassification and non-response bias. Fourth, we performed the screening with OS and cancer-related genes identified in a systematic review. This hypothesis-based gene-specific approach has a higher statistical power than an epigenome-wide screening approach. Lastly, the 8-isoprostane molecule has proven long-term stability in frozen urine samples and is a reliable biomarker for OS [34]. Nevertheless, DNA methylation at cg25365794 (ALOXE3 gene) and cg08862778 (MTOR gene) may be useful in the future as biomarkers for long-term OS exposure because they may have a higher intra-individual stability time than urinary concentrations of 8-isoprostane.
However, several limitations of our analysis should be taken into account when interpreting the results. First, DNA methylation levels vary across tissue types [35] and whole blood DNA methylation can only reflect the overall methylation levels in leukocytes. Second, screening and validation sets were obtained from the same study population. Other studies are needed to corroborate our findings for the ALOXE3 and MTOR genes, to conduct gene expression analyses in multiple CpG sites in the promotor/CpG islands of these genes, and to find further CpG sites with OS-related DNA methylation. This may provide further insights into the mechanisms of OS-related cancer development and aging.
Conclusion
In this population-based cohort, DNA methylation at two CpG sites was associated with urinary 8-isoprostane levels. DNA methylation alterations at the identified CpG sites were associated with specific cancer outcomes. While the association between urinary 8-isoprostane levels and DNA methylation at the ALOXE3 gene may not be causal, the findings for MTOR gene methylation are biologically plausible. There might be a pathway of ROS-mediated PI3K/AKT/mTOR activation to breast cancer that is in part controlled by DNA demethylation in the promoter region of the MTOR gene.
References
Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8:1865–79.
Sies H, Berndt C, Jones DP. Oxidative stress. Annu Rev Biochem. 2017;86:715–48.
Yan L-J. Positive oxidative stress in aging and aging-related disease tolerance. Redox Biol. 2014;2:165–9.
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84.
Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol. 2011;7:504–11.
Milne GL, Dai Q, Roberts LJ 2nd. The isoprostanes--25 years later. Biochim Biophys Act. 2015;1851:433–45.
Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5:9–19.
Gào X, Brenner H, Holleczek B, et al. Urinary 8-isoprostane levels and occurrence of lung, colorectal, prostate, breast and overall cancer: results from a large, population-based cohort study with 14 years of follow-up. Free Radic Biol Med. 2018;123:20–6.
Yuan JM, Carmella SG, Wang R, et al. Relationship of the oxidative damage biomarker 8-epi-prostaglandin F2alpha to risk of lung cancer development in the Shanghai Cohort Study. Carcinogenesis. 2018;39:948-54.
Franco R, Schoneveld O, Georgakilas AG, Panayiotidis MI. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008;266:6–11.
Mahalingaiah PKS, Ponnusamy L, Singh KP. Oxidative stress-induced epigenetic changes associated with malignant transformation of human kidney epithelial cells. Oncotarget. 2017;8:11127–43.
Afanas'ev I. New nucleophilic mechanisms of ros-dependent epigenetic modifications: comparison of aging and cancer. Aging Dis. 2014;5:52–62.
Rang FJ, Boonstra J. Causes and consequences of age-related changes in DNA methylation: a role for ROS? Biol. 2014;3:403–25.
Niu Y, DesMarais TL, Tong Z, Yao Y, Costa M. Oxidative stress alters global histone modification and DNA methylation. Free Radic Biol Med. 2015;82:22–8.
Schöttker B, Zhang Y, Heiss JA, et al. Discovery of a novel epigenetic cancer marker related to the oxidative status of human blood. Genes Chromosomes Cancer. 2015;54:583–94.
Gao T, Joyce BT, Liu L, et al. DNA methylation of oxidative stress genes and cancer risk in the Normative Aging Study. Am J Cancer Res. 2016;6:553–61.
Löw M, Stegmaier C, Ziegler H, Rothenbacher D, Brenner H. Epidemiological investigations of the chances of preventing, recognizing early and optimally treating chronic diseases in an elderly population (ESTHER study). Dtsch Med Wochenschr. 2004;129:2643–7.
Schöttker B, Haug U, Schomburg L, et al. Strong associations of 25-hydroxyvitamin D concentrations with all-cause, cardiovascular, cancer, and respiratory disease mortality in a large cohort study. Am J Clin Nutr. 2013;97:782–93.
Zhang Y, Saum KU, Schöttker B, Holleczek B, Brenner H. Methylomic survival predictors, frailty, and mortality. Aging. 2018;10:339–57.
Zhang Y, Wilson R, Heiss J, et al. DNA methylation signatures in peripheral blood strongly predict all-cause mortality. Nat Commun. 2017;8:14617.
Gào X, Schöttker B. Reduction-oxidation pathways involved in cancer development: a systematic review of literature reviews. Oncotarget. 2017;8:51888–906.
Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
Zheng Y, Brash AR. On the role of molecular oxygen in lipoxygenase activation: comparison and contrast of epidermal lipoxygenase-3 with soybean lipoxygenase-1. J Biol Chem. 2010;285:39876–87.
Krieg P, Rosenberger S, de Juanes S, et al. Aloxe3 knockout mice reveal a function of epidermal lipoxygenase-3 as hepoxilin synthase and its pivotal role in barrier formation. J Invest Dermatol. 2013;133:172–80.
Krieg P, Marks F, Furstenberger G. A gene cluster encoding human epidermis-type lipoxygenases at chromosome 17p13.1: cloning, physical mapping, and expression. Genomics. 2001;73:323–30.
Andreotti G, Koutros S, Berndt SI, et al. The interaction between pesticide use and genetic variants involved in lipid metabolism on prostate cancer risk. J Cancer Epidemiol. 2012;2012:358076.
Heberle AM, Prentzell MT, van Eunen K, Bakker BM, Grellscheid SN, Thedieck K. Molecular mechanisms of mTOR regulation by stress. Mol Cell Oncol. 2015;2:e970489.
Kim LC, Cook RS, Chen J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene. 2017;36:2191–201.
Lipton JO, Sahin M. The neurology of mTOR. Neuron. 2014;84:275–91.
Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–94.
Wang X, Proud CG. mTORC2 is a tyrosine kinase. Cell Res. 2016;26:1–2.
Sendur MA, Zengin N, Aksoy S, Altundag K. Everolimus: a new hope for patients with breast cancer. Curr Med Res Opin. 2014;30:75–87.
Tang Q, Holland-Letz T, Slynko A, et al. DNA methylation array analysis identifies breast cancer associated RPTOR, MGRN1 and RAPSN hypomethylation in peripheral blood DNA. Oncotarget. 2016;7:64191–202.
Yan W, Byrd GD, Ogden MW. Quantitation of isoprostane isomers in human urine from smokers and nonsmokers by LC-MS/MS. J Lipid Res. 2007;48:1607–17.
Christensen BC, Houseman EA, Marsit CJ, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5:e1000602.
Acknowledgements
The authors thank the study participants and their general practitioners as well as laboratory and administrative staff of the ESTHER study team. The authors gratefully acknowledge contributions of DKFZ Genomics and Proteomics Core Facility in the processing of DNA samples and performing the laboratory work.
Funding
This study was funded by a grant from the German Research Foundation (grant No. SCHO 1545/3-1) and by China Scholarship Council (grant No. 201506010268 to Xīn Gào). The ESTHER study was funded by grants from the Saarland state Ministry for Social Affairs, Health, Women and Family Affairs (Saarbrücken, Germany), the Baden-Württemberg state Ministry of Science, Research and Arts (Stuttgart, Germany), the Federal Ministry of Education and Research (Berlin, Germany) and the Federal Ministry of Family Affairs, Senior Citizens, Women and Youth (Berlin, Germany).
Availability of data and materials
The datasets generated and/or analyzed during the current study are not made publicly available due to ethical and data security requirements but can be made available for researchers on the basis of a research proposal (to be submitted to the corresponding author).
Author information
Authors and Affiliations
Contributions
XG and BS wrote the research proposal and BS supervised the project. HB, BS, and YZ contributed to data collection for the ESTHER study. BH coordinated the cancer registry data collection. XG performed the statistical analyses under supervision of YZ and BS. XG wrote the first draft of the manuscript and BS edited it. All authors contributed to the interpretation of the results and approved and the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The ESTHER Study has been approved by the ethics committees of the Medical Faculty of the University of Heidelberg and the Medical Association of Saarland and is being conducted in accordance with the declaration of Helsinki. Written informed consent was obtained from all study participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. 519 genes coding for proteins involved in pathways from oxidative stress to cancer. Table S2. Top 10 CpG sites with respect to associations with 8-isoprostane concentrations in the derivation set. Figure S1. Distributions of the methylation levels of the selected CpG sites across subsets. Figure S2. Scatter plots showing linear associations between DNA methylation at two selected CpG sites and 8-isoprostane levels (log-transformation) across subsets. (DOCX 744 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Gào, X., Zhang, Y., Burwinkel, B. et al. The associations of DNA methylation alterations in oxidative stress-related genes with cancer incidence and mortality outcomes: a population-based cohort study. Clin Epigenet 11, 14 (2019). https://doi.org/10.1186/s13148-018-0604-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-018-0604-y