
Abstract
Sirtuins are NAD+-dependent histone deacetylases regulating important metabolic pathways in prokaryotes and eukaryotes and are involved in many biological processes such as cell survival, senescence, proliferation, apoptosis, DNA repair, cell metabolism, and caloric restriction. The seven members of this family of enzymes are considered potential targets for the treatment of human pathologies including neurodegenerative diseases, cardiovascular diseases, and cancer. Furthermore, recent interest focusing on sirtuin modulators as epigenetic players in the regulation of fundamental biological pathways has prompted increased efforts to discover new small molecules able to modify sirtuin activity. Here, we review the role, mechanism of action, and biological function of the seven sirtuins, as well as their inhibitors and activators.
Similar content being viewed by others
Background
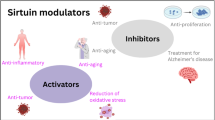
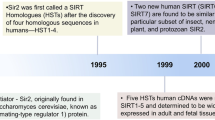
The mammalian sirtuin (SIRT) family, evolutionally conserved proteins belonging to class III histone deacetylases (HDACs), comprises seven members. SIRTs share a NAD+-binding catalytic domain and may act specifically on different substrates depending on the biological processes in which they are involved (Table 1). SIRTs differ in sequence and length in both their N- and C-terminal domains, partially explaining their different localization and functions. SIRTs can catalyze both deacetylation and ADP-ribosylation [1]. Their best-characterized activity is NAD+-dependent lysine deacetylation, but recent studies demonstrated that some SIRTs also remove other acyl groups such as succinyl, malonyl, glutaryl, and long-chain fatty acyl groups [2, 3] (Table 1). The last decade has seen growing interest in their association with and involvement in different pathologies, such as (but not restricted to) cancer and neurodegenerative diseases. Increasing evidences supporting the potential use of SIRT inhibitors (SIRTi) for the treatment of cancer, HIV infection, and muscular diseases and of SIRT activators (SIRTa) for age-related disorders have led to the identification of many SIRT modulators over recent years, mainly through chemical library screening and catalytic mechanism-based design approaches, often combined with structure-activity relationship (SAR) investigations. Here, we provide a summary of the most important SIRT activities and SIRT modulators, their latest analogs, and novel chemotypes (Tables 2 and 3).
Sirtuins in physiology and pathology
SIRT1 was the first SIRT family member to be discovered and is still the most studied. Its involvement in many neuronal processes [4, 5] prompted further investigations into its role in neurological disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD) [6–8]. SIRT1 exerts a neuroprotective action and is involved in survival, neuropathology, and the expression of brain-derived neurotrophic factor. In a mouse model of HD, SIRT1 knockout results in exacerbation of the pathology, while its re-expression displays neuroprotective effects [9]. A correlation between nicotinamide phosphoribosyltransferase (NAMPT) and SIRT1 was recently shown. NAMPT is a therapeutic target against ischemic stroke, acting in vascular repair and neurogenesis. SIRT1-mediated deacetylation of NAMPT at K53 increased its activity and secretion [10]. The first observations linking SIRT1 to tumorigenesis came from two studies showing p53 deacetylation and inhibition [11, 12], promoting cancer cell death. In tumorigenesis, SIRT1 seems to play a contradictory role, acting as both a tumor promoter and tumor suppressor (by inhibiting oncogenes and oncoproteins, similar to survivin) [13, 14]. SIRT1 regulates many tumor suppressors and DNA repair genes [15–18]. SIRT1 upregulation is described in many human malignancies [19]. Although its role in cancer remains contradictory [20], recent studies showed that patients expressing high levels of SIRT1 had a higher chance of being resistant to chemotherapy than patients with low SIRT1 expression [21, 22]. One report proposed a role for SIRT1 in melanoma and suggested the use of inhibitors (tenovins, EX-527, and sirtinol), either alone or in combination. These inhibitors act on a variety of SIRTs, strengthening the idea that the concomitant inhibition of several SIRTs may contribute to a reduction in malignant growth [23]. SIRT2, the second member of the SIRT family, seems to exert a neurodegenerative action in neurological disease [24]. Indeed, pharmacological or genetic inhibition of SIRT2 blocks of αSyn-mediated toxicity [25]. SIRT2 is involved in apoptosis control through p53 process regulation. Several studies confirm the role of SIRT2 in the control of cell cycle progression at many levels. It was demonstrated that it is a checkpoint for metaphase/anaphase processes and G2/M transition [26, 27]. Recent studies indicated that SIRT2 might also act as a tumor promoter or tumor suppressor in tumorigenesis, likely displaying a regulatory function [28, 29]. Moreover studies about SIRT2 have shown its involvement in metabolism processes, like adipogenesis [30]. SIRT2 is capable to deacetylate the Glucokinase (GCK), an essential enzyme for maintaining homeostasis of glucose regulated by the binding of the glucokinase regulatory protein (GKRP). Acetylated GKRP is connected to diabetes mellitus [31]. Of the three mitochondrial SIRTs (SIRT3, 4, and 5), some evidence correlates SIRT3 with neurodegenerative disease. SIRT3 protects neurons in the cochlea against oxidative damage during calorie restriction [32] and in response to stress-regulating mitochondrial antioxidant manganese superoxide dismutase (MnSOD) in microglia [33]. A number of studies also hypothesize its involvement in cancer progression. SIRT3 mainly inhibits mitochondrial ROS production but also deacetylates and activates many mitochondrial proteins, regulating proliferation, differentiation, and survival [34, 35]. Recent data suggest that SIRT3 acts as a tumor suppressor by inhibiting glycolysis metabolism after the deacetylation and activation of pyruvate dehydrogenase; thus, also the role of SIRT3 in cancer is debatable. Much less is known about the remaining SIRTs. Unlike other family members, SIRT4 lacks NAD+-dependent deacetylase activity but exerts ADP-ribosyltransferase activity on histones. SIRT4 is induced by DNA damage including chemotherapy and γ-irradiation and is capable of arresting the cell cycle by inhibiting mitochondrial glutamine metabolism. SIRT4 inhibits proliferation, invasion, and migration in colorectal cancer cells, and its low expression is correlated with a worse prognosis [36]. SIRT5 is reported to interact with carbamoyl phosphate synthetase 1 (CPS1), and it is deacetylated by SIRT5. The role of SIRT5 in carcinogenesis remains unclear, but a recent study showed its overexpression in non-small cell lung cancer tissues as a marker of low survival [37]. SIRT6 was reported to display actions controlling cellular homeostasis, DNA repair, telomere maintenance, and metabolism, acting as an epigenetic guardian for cellular differentiation [38]. Recent studies have highlighted SIRT7 as a promising target for epigenetic cancer therapy. SIRT7 catalyzes selective deacetylation of H3K18, an emerging epigenetic biomarker of aggressive tumors, controlling many tumor suppressor genes. High expression of SIRT7 has in fact been associated with aggressive cancers and low survival, whereas its depletion leads to a less aggressive phenotype [39]. A recent report described the use of chlorpromazine to inhibit cell cycle progression in rat glioma cells and induce autophagic cell death. The authors demonstrate that an FDA-approved drug used to treat schizophrenia and bipolar disorder is able to increase p53 Lys382 acetylation through a mechanism of SIRT1 inhibition [40].
SIRT inhibition—small molecules
Many SIRT modulators, used alone or in combination with other epigenetic modulators or known drugs, have been described as having beneficial effects against neurodegeneration and cancer [41]. Identified in 2001 from a yeast-based screening for inhibitors of Sir2, splitomicin (1, Table 2), despite being substantially inactive against hSIRTs, has been the starting point for the development of several SIRT1/2 inhibitors [42]. Among them, the compound with a phenyl ring on 2-position and a bromine on 8-position indicated as HR-73 (2, Table 2) is a single-digit micromolar SIRT1i that inhibits HIV transcription through affecting the Tat protein acetylation [43]. In a series of splitomicin analogs carrying a phenyl ring on 3-position and different substituents on 8-position, compounds 3 and 4 (Table 2), respectively, revealed to be low micromolar SIRT2i able to exert antiproliferative effects and cause α-tubulin hyperacetylation in MCF-7 cells [44]. Discovered in 2005, EX-527 (selisistat, 5, Table 2) was the first potent, selective (over SIRT2/3), and cell-permeable SIRT1i [45, 46]. Despite increasing p53 acetylation, EX-527 has no major effect on viability and proliferation in various tumors [47]. EX-527 is being developed for HD. A phase II clinical study recently demonstrated that EX-527 is safe and well tolerated in HD patients at plasma concentrations, providing benefit in non-clinical HD models [48]. Recently, EX-527 was also found to block the amplification of human papillomavirus via a SIRT1 inhibition-dependent mechanism [49]. Identified in 2006, cambinol (6, Table 2) is a moderate SIRT1/2i that induces hyperacetylation of p53, α-tubulin, FOXO3a, and Ku70 in NCI-H460 and HeLa cancer cells; promotes apoptosis in BCL-6-expressing Burkitt lymphoma cells; and reduces tumor growth in a xenograft model [50]. It is also endowed with anticancer effects on hepatocellular carcinoma tumor models in vitro and in vivo [51] and reduces neuroblastoma formation in N-Myc transgenic mice [52]. These promising results stimulated various optimization efforts, leading to the identification of derivatives/analogs with increased potency/selectivity for either SIRT1 or SIRT2 [53–55]. Discovered through a focused library screening, AGK2 (7, Table 2) is a single-digit micromolar SIRT2i selective over SIRT1/3. Able to inhibit the deacetylation of α-tubulin in HeLa cells, AGK2 rescues dopaminergic neurons from α-synuclein toxicity and protects against PD in different PD models [25]. Likely through SIRT2 inhibition, AGK2 also induces caspase-3-dependent death in glioma cells [56]. A cell-based screen designed to detect p53 activators identified tenovin-1 (8, Table 2) and its water-soluble analog tenovin-6 (9, Table 2) as SIRTi. Tenovin-6 is a micromolar SIRT1-3i that increases the level of p53K382ac; shows cytotoxic effects on melanoma cells, delaying the growth of ARN8-derived xenograft tumors [57]; and induces apoptosis in gastric cancer cells [58] and in chronic myeloid leukemia cells, decelerating the disease progression in mice models [59]. Discovered in 2001 by a high-throughput cell-based screening, sirtinol (10, Table 2) is a micromolar ySir2 and hSIRT1/2 inhibitor endowed with various anticancer activities [60]. It induces senescence-like growth arrest in human MCF-7 and H1299 cells [61]; inhibits the growth of PC3, DU145, and HeLa cells, enhancing their chemosensitivity to cisplatin and camptothecin [62, 63]; and promotes significant growth inhibition or apoptosis in cells from adult T cell leukemia-lymphoma (ATL) [64]. Salermide (11, Table 2), which resulted from the med-chem optimization of sirtinol, is a moderate SIRT1/2i that induces cancer-specific pro-apoptotic effects on different tumor cells (mainly MOLT4, KG1A, SW480, and Raji) through reactivation of pro-apoptotic genes (CASP8, TNF, TNFRSF10B, and PUMA) previously repressed by SIRT1-mediated H4K16ac deacetylation [65] and through the upregulation of DR5 [66]. Salermide and its analogs also show potent antiproliferative effects on cancer stem cells [67] and exert cell protection effects on a C. elegans model of muscular dystrophy [68]. In infectious diseases (Schistosoma mansoni), salermide was the most potent among the tested SIRTi in inducing apoptosis and death of schistosomula, in separation of adult worm pairs and in reduction in egg laying, through inhibition of the S. mansoni specific sirtuin [69]. Identified in 2010 as result of cambinol manipulation, MC2141 (12, Table 2) is the prototype of a series of benzodeazaoxaflavins active as low micromolar SIRT1/2i. MC2141 displays significant pro-apoptotic and antiproliferative properties in different cancer cell lines, including cancer stem cells [70, 71]. Identified in 2012 in a virtual screening for inhibitors of the p53-MDM2/MDMX interaction, inauhzin (13, Table 2) is a (sub)micromolar SIRT1i selective over SIRT2/3 able to exert antiproliferative and tumor-specific pro-apoptotic effects on various cancer lines by activating and stabilizing p53. Inauhzin represses the growth of xenograft tumors derived from p53-harboring H460 and HCT116 cells [72]. The most potent SIRTi reported to date are thieno[3,2-d]pyrimidine-6-carboxamides that resulted from a SAR analysis of the hit compounds deriving from a DNA-encoded small molecule library screen [73]. Although the most potent compound in the series (14, Table 2) is active in the single-digit nanomolar range against SIRT1–3 and its binding mode has been elucidated by X-ray crystallography, no biological activities are currently known for these compounds. Very recently, SirReal2 (15, Table 2) was reported as a submicromolar SIRT2i with >1000-fold selectivity over SIRT1/3/4/5/6 [74]. Its potency and selectivity, as revealed by X-ray crystallography, are based on a ligand-induced structural rearrangement of the SIRT2 active site revealing a previously unexploited binding pocket. The SIRT2 inhibition capability of SirReal2 has been confirmed in HeLa cells by the induction of α-tubulin hyperacetylation and the destabilization of the SIRT2 substrate BubR1. A subsequent SAR investigation on the prototype led to the development of some derivatives (16–20, Table 2) with improved SIRT2i potency [75].
An elegant fragment-based approach inspired by the structures of the SIRTi suramin and nicotinamide recently identified a nanomolar SIRT2-selective (over SIRT1/3) inhibitor (21, Table 2), which is able to induce clear time-dependent and dose-dependent hyperacetylation of α-tubulin in MCF-7 cells and shows cytotoxic effects on some cancer cell lines [76]. In recent years, a series of chroman-4-one derivatives as low micromolar SIRT2i selective over SIRT1/3 has been reported [77, 78]. Among them, compounds 22 and 23 (Table 2) are the most attractive in terms of their overall pharmacological profile as they display good dose-dependent α-tubulin hyperacetylation and significant antiproliferative effects on cancer cells (MCF-7 and A549). From the screening of a panel of polyglutamine aggregate inhibitors, AK-7 (24, Table 2) was identified in 2011 as a brain-permeable micromolar SIRT2 selective (over SIRT1/3) inhibitor able to reduce neuronal cholesterol biosynthesis [79]. In addition, AK-7 displays other SIRT2 inhibition-dependent neuroprotective effects on various models of HD [80] and PD [81], supporting the development of SIRT2i as potential therapeutics for these disorders. Very recently, by high-throughput screening employing self-assembled monolayer desorption/ionization mass spectrometry, the first submicromolar small molecule inhibitor of SIRT3, SDX-437 (25, Table 2), was identified. SDX-437 is >100-fold selective over SIRT1 and could be a useful tool for studying SIRT3 biology and a promising lead for future med-chem optimization campaigns [82]. By a high-throughput molecular docking screen, compound 26 (Table 2) was recently identified as the first micromolar small molecule inhibitor of SIRT6 with >eightfold selectivity over SIRT1/2 [83]. Although this molecule needs optimization, it can be considered as a promising lead for the functional annotation of SIRT6 and the development of potential SIRT6-based therapeutics due to its ability to induce hyperacetylation of the SIRT6 substrate H3K9, to increase glucose uptake through the upregulation of GLUT-1, and to reduce TNF-α secretion (BxPC-3 cells).
SIRT inhibition—peptides and pseudopeptides
The first peptidic SIRTi was developed by replacing the N ε-acetyl-lysine with a thioacetylated residue and by using the C-terminus of p53 protein (H2N-KKGQSTSRH(ThAcK)LMFKTEG-COOH) as a substrate peptide template [84]. Based on kinetic studies, this thioacetylated peptide was able to form an intermediate tighter than the acetylated peptide [84–86], showing a strong SIRT1 inhibition (IC50 ~ 2 μM), and was also active against SIRT2/3 (IC50 ~ 2 and 67 μM, respectively) [87]. More thioacetylated peptide inhibitors were developed from other SIRT substrates such as human α-tubulin and acetyl-coenzyme A synthetase 2 (AceCS2) [87, 88]. The α-tubulin [85–93]-based H2N-MPSD(ThAcK)TIGG-COOH inhibited SIRT2 at a low micromolar level (IC50 ~ 11 μM), whereas it was less potent against SIRT1/3. The thioacetylated AceCS2 (633–652) peptide showed low micromolar inhibition of SIRT1–3 (IC50 ~ 0.9, 4, and 5 μM, respectively) [87]. Thioacetylated α-tubulin and p53-based tri-, tetra-, and pentapeptides were developed in 2009 [88]. Several peptides had submicromolar IC50s for SIRT1, such as tripeptide K(ThAcK)L (0.57 μM) and pentapeptide HK(ThAcK)LM (0.18 μM). The most potent inhibitor for SIRT2 was the pentapeptide HK(ThAcK)AM (IC50 ~ 3.8 μM). The first small pseudopeptidic SIRTi was reported in 2009 [89]. The most potent compound, NCS-12k (27, Table 2), showed an IC50 of 3.9 μM for SIRT1. Further modifications produced the cell-active Cbz-Lys(ThAc)-NH-Ph (28, Table 2) with an IC50 of 2.7 μM for SIRT1, 23 μM for SIRT2, but interestingly >100 μM for SIRT3 [90]. Among other N ε-thioacetylated pseudopeptide inhibitors developed to improve potency, selectivity, and cell permeability, compounds 29 and 30 (Table 2) were found to be micromolar inhibitors of SIRT1-3 and showed antiproliferative effects on cancer cells [91, 92]. Some pseudopeptidic inhibitors were also tested against SIRT6, but potency was lower [93]. Besides the thioacetyl group, several other lysine N ε-modifications (such as propionyl, butyryl, α-hydroxyacetyl, monofluoroacetyl, homocitrulline, homoarginine, difluoroacetyl, acetimidoyl, methanesulfonyl, selenoacetyl, and trifluoroacetyl groups) have been reported [85, 86, 93–95]. Among the carbonyl-containing N ε-modifications studied on the sequence Ac-AKA-OH, the most potent were N ε-3,3-dimethylacryl (31, Table 2) and N ε-isovaleryl (32, Table 2) with IC50s in the micromolar range against SIRT1/2, while N ε-isothiovaleryl (33, Table 2) improved potency giving SIRT1/2 IC50s in the (sub)micromolar range [95].
SIRT activators
The natural polyphenol resveratrol (34, Table 3) was the first SIRT1a described. Its administration extends lifespan in yeast, Caenorhabditis elegans, Drosophila, fishes, and bees [96–99]. Mice treated with resveratrol display improved mitochondrial functions and protection against high-fat diet-induced obesity [100], while in obese mice, treatment with resveratrol leads to increased healthspan and lifespan [101]. Identified in 2007 by high-throughput screening as selective SIRT1a more potent than resveratrol, SRT compounds (35–37, Table 3) reproduce most of resveratrol beneficial effects in vivo. In diet-induced and genetically obese (Lepob/ob) mice, compounds 35–37 (Table 3) improve insulin sensitivity, lower plasma glucose, and increase mitochondrial capacity. Moreover, in Zucker (fa/fa) rats, the same compounds improve whole-body glucose homeostasis and insulin sensitivity [95]. Due to their very promising activities, some of the most potent SRTs are currently in clinical trials for the treatment of different age-related diseases [102–106]. The most potent scaffolds proposed as sirtuin-activating in terms of SIRT1 activation (EC1.5 values in the submicromolar range) and the most effective in the treatment of age-related conditions (obesity, metabolic and cardiovascular disorders, inflammatory/autoimmune diseases, neurodegeneration, and cancer) are pyrido[3,2-b][1, 4]oxazocines (38, Table 3) [104–106]; oxazolo[4,5-b]pyridines and benzo[d]imidazoles (39–41, Table 3) [104–107]; thiazolopyridines (42–44, Table 3) [104–106, 108]; and azabenzimidazoles (45, Table 3) [104–106, 109]. Over the past years, it has been highly debated the effective SIRT1 activation by resveratrol and SRT compounds, because only the use in the enzyme assays of aminomethylcoumarin (AMC)- or carboxytetramethylrhodamine (TAMRA)-labeled substrates allowed to see activation, whereas in the presence of natural substrates no activation was observed [110–112]. For this reason, it was questioned if the action of resveratrol and SRT compounds on SIRT1 is the result of a direct activation, or if rather it arises from the indirect modulation of other pathways, such as AMPK activation and/or PDE inhibition [113]. Recently, direct SIRT1 activation has been confirmed, and it has been shown that there are subtle structural and positional requirements to detect SIRT1 activation with some of its natural substrates (e.g., FOXO3a and PGC-1α) [104, 114]. Moreover, E230K or E230A SIRT1 mutation abolishes enzyme activation as well as binding of SRT compounds, suggesting an assisted allosteric activation mechanism in which the activators bind and stabilize the enzyme-substrate complex promoting the deacetylation reaction [104]. Lastly, the crystal structure of a SRT compound bound to an engineered minimally functional hSIRT1 has been reported, unambiguously confirming the direct allosteric activation of SIRT1 by small molecules [115]. The 1,4-dihydropyridine (DHP) scaffold with general formula 46 (Table 3) has been described as a new chemotype for SIRT activation. Selected DHPs induced hypoacetylation of α-tubulin in U937 cells, high NO release in HaCat cells, and ameliorated skin repair in a mouse model of wound healing. In myoblasts, they improved mitochondrial density and functions through activation of the SIRT1/AMPK axis. A water-soluble analog displayed antiproliferative action and increased H4K16ac deacetylation in a panel of cancer cells at 8–35 μM [116–118]. Very recently, the cardiac anti-hypertrophic effects of the natural compound honokiol (47, Table 3) were reported to depend on the pharmacological activation (increased expression and activity) of SIRT3 [119].
Conclusions
A critical analysis of current knowledge and recent discoveries clearly indicates that SIRT modulation is beneficial against several diseases including cancer and neurodegeneration. Chronic diseases, such as obesity and diabetes, might also likely benefit from SIRT targeting, but further investigation is needed. The ubiquitous expression of some SIRTs, together with the fact that many studies primarily investigated SIRT1 only, and not all the other family members, still leaves room for some confusion. Currently, for example, activation or inhibition of SIRTs (even the same SIRT) is described as beneficial for pathologies such as neurodegeneration. Despite the apparent contradiction, this is theoretically possible when taking into account the role of SIRT-containing complexes and not just the catalytic domain of the single enzyme. Furthermore, both the chemistry and biomedical characterization of SIRTa are at a much earlier stage of development than SIRTi, thus suggesting that new alternatives may arise in the near future. Finally, the potential selective modulation of SIRT family members will represent a promising area provided that new selective molecules can be designed.
References
Carafa V, Nebbioso A, Altucci L. Sirtuins and disease: the road ahead. Front Pharmacol. 2012;3:4. doi:10.3389/fphar.2012.00004.
Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014;19(4):605–17. doi:10.1016/j.cmet.2014.03.014.
Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334(6057):806–9. doi:10.1126/science.1207861.
Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305(5686):1010–3. doi:10.1126/science.1098014.
Li XH, Chen C, Tu Y, Sun HT, Zhao ML, Cheng SX, et al. Sirt1 promotes axonogenesis by deacetylation of Akt and inactivation of GSK3. Mol Neurobiol. 2013;48(3):490–9. doi:10.1007/s12035-013-8437-3.
Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006;281(31):21745–54. doi:10.1074/jbc.M602909200.
Donmez G, Arun A, Chung CY, McLean PJ, Lindquist S, Guarente L. SIRT1 protects against alpha-synuclein aggregation by activating molecular chaperones. J Neurosci. 2012;32(1):124–32. doi:10.1523/JNEUROSCI.3442-11.2012.
Jeong JK, Moon MH, Lee YJ, Seol JW, Park SY. Autophagy induced by the class III histone deacetylase Sirt1 prevents prion peptide neurotoxicity. Neurobiol Aging. 2013;34(1):146–56. doi:10.1016/j.neurobiolaging.2012.04.002.
Jeong H, Cohen DE, Cui L, Supinski A, Savas JN, Mazzulli JR, et al. Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat Med. 2012;18(1):159–65. doi:10.1038/nm.2559.
Yoon MJ, Yoshida M, Johnson S, Takikawa A, Usui I, Tobe K, et al. SIRT1-mediated eNAMPT secretion from adipose tissue regulates hypothalamic NAD+ and function in mice. Cell Metab. 2015;21(5):706–17. doi:10.1016/j.cmet.2015.04.002.
Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107(2):137–48.
Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107(2):149–59.
Deng CX. SIRT1, is it a tumor promoter or tumor suppressor? Int J Biol Sci. 2009;5(2):147–52.
Bosch-Presegue L, Vaquero A. The dual role of sirtuins in cancer. Genes Cancer. 2011;2(6):648–62. doi:10.1177/1947601911417862.
Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305(5682):390–2. doi:10.1126/science.1099196.
Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, et al. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116(4):551–63.
Pruitt K, Zinn RL, Ohm JE, McGarvey KM, Kang SH, Watkins DN, et al. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2006;2(3):e40. doi:10.1371/journal.pgen.0020040.
Dai JM, Wang ZY, Sun DC, Lin RX, Wang SQ. SIRT1 interacts with p73 and suppresses p73-dependent transcriptional activity. J Cell Physiol. 2007;210(1):161–6. doi:10.1002/jcp.20831.
Derr RS, van Hoesel AQ, Benard A, Goossens-Beumer IJ, Sajet A, Dekker-Ensink NG, et al. High nuclear expression levels of histone-modifying enzymes LSD1, HDAC2 and SIRT1 in tumor cells correlate with decreased survival and increased relapse in breast cancer patients. BMC Cancer. 2014;14:604. doi:10.1186/1471-2407-14-604.
Chen X, Sun K, Jiao S, Cai N, Zhao X, Zou H, et al. High levels of SIRT1 expression enhance tumorigenesis and associate with a poor prognosis of colorectal carcinoma patients. Sci Rep. 2014;4:7481. doi:10.1038/srep07481.
Zhang T, Rong N, Chen J, Zou C, Jing H, Zhu X, et al. SIRT1 expression is associated with the chemotherapy response and prognosis of patients with advanced NSCLC. PLoS ONE. 2013;8(11):e79162. doi:10.1371/journal.pone.0079162.
Li C, Wang L, Zheng L, Zhan X, Xu B, Jiang J, et al. SIRT1 expression is associated with poor prognosis of lung adenocarcinoma. Onco Targets Ther. 2015;8:977–84. doi:10.2147/OTT.S82378.
Wilking MJ, Singh CK, Nihal M, Ndiaye MA, Ahmad N. Sirtuin deacetylases: a new target for melanoma management. Cell Cycle. 2014;13(18):2821–6. doi:10.4161/15384101.2014.949085.
Harting K, Knoll B. SIRT2-mediated protein deacetylation: an emerging key regulator in brain physiology and pathology. Eur J Cell Biol. 2010;89(2-3):262–9. doi:10.1016/j.ejcb.2009.11.006.
Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science. 2007;317(5837):516–9. doi:10.1126/science.1143780.
Serrano L, Martinez-Redondo P, Marazuela-Duque A, Vazquez BN, Dooley SJ, Voigt P, et al. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 2013;27(6):639–53. doi:10.1101/gad.211342.112.
Inoue T, Hiratsuka M, Osaki M, Yamada H, Kishimoto I, Yamaguchi S, et al. SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene. 2007;26(7):945–57. doi:10.1038/sj.onc.1209857.
Li Z, Xie QR, Chen Z, Lu S, Xia W. Regulation of SIRT2 levels for human non-small cell lung cancer therapy. Lung Cancer. 2013;82(1):9–15. doi:10.1016/j.lungcan.2013.05.013.
McGlynn LM, Zino S, MacDonald AI, Curle J, Reilly JE, Mohammed ZM, et al. SIRT2: tumour suppressor or tumour promoter in operable breast cancer? Eur J Cancer. 2014;50(2):290–301. doi:10.1016/j.ejca.2013.10.005.
Jing E, Gesta S, Kahn CR. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007;6(2):105–14. doi:10.1016/j.cmet.2007.07.003.
Park JM, Kim TH, Jo SH, Kim MY, Ahn YH. Acetylation of glucokinase regulatory protein decreases glucose metabolism by suppressing glucokinase activity. Sci Rep. 2015;5:17395. doi:10.1038/srep17395.
Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143(5):802–12. doi:10.1016/j.cell.2010.10.002.
Rangarajan P, Karthikeyan A, Lu J, Ling EA, Dheen ST. Sirtuin 3 regulates Foxo3a-mediated antioxidant pathway in microglia. Neuroscience. 2015;311:398–414. doi:10.1016/j.neuroscience.2015.10.048.
Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell. 2010;40(6):893–904. doi:10.1016/j.molcel.2010.12.013.
Finley LW, Haas W, Desquiret-Dumas V, Wallace DC, Procaccio V, Gygi SP, et al. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS ONE. 2011;6(8):e23295. doi:10.1371/journal.pone.0023295.
Miyo M, Yamamoto H, Konno M, Colvin H, Nishida N, Koseki J, et al. Tumour-suppressive function of SIRT4 in human colorectal cancer. Br J Cancer. 2015;113(3):492–9. doi:10.1038/bjc.2015.226.
Lu W, Zuo Y, Feng Y, Zhang M. SIRT5 facilitates cancer cell growth and drug resistance in non-small cell lung cancer. Tumour Biol. 2014;35(11):10699–705. doi:10.1007/s13277-014-2372-4.
Etchegaray JP, Chavez L, Huang Y, Ross KN, Choi J, Martinez-Pastor B, et al. The histone deacetylase SIRT6 controls embryonic stem cell fate via TET-mediated production of 5-hydroxymethylcytosine. Nat Cell Biol. 2015;17(5):545–57. doi:10.1038/ncb3147.
Barber MF, Michishita-Kioi E, Xi Y, Tasselli L, Kioi M, Moqtaderi Z, et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature. 2012;487(7405):114–8. doi:10.1038/nature11043.
Lee WY, Lee WT, Cheng CH, Chen KC, Chou CM, Chung CH, et al. Repositioning antipsychotic chlorpromazine for treating colorectal cancer by inhibiting sirtuin 1. Oncotarget. 2015;6(29):27580–95. doi:10.18632/oncotarget.4768.
Villalba JM, Alcain FJ. Sirtuin activators and inhibitors. Biofactors. 2012;38(5):349–59. doi:10.1002/biof.1032.
Bedalov A, Gatbonton T, Irvine WP, Gottschling DE, Simon JA. Identification of a small molecule inhibitor of Sir2p. Proc Natl Acad Sci U S A. 2001;98(26):15113–8. doi:10.1073/pnas.261574398.
Pagans S, Pedal A, North BJ, Kaehlcke K, Marshall BL, Dorr A, et al. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 2005;3(2):e41. doi:10.1371/journal.pbio.0030041.
Neugebauer RC, Uchiechowska U, Meier R, Hruby H, Valkov V, Verdin E, et al. Structure-activity studies on splitomicin derivatives as sirtuin inhibitors and computational prediction of binding mode. J Med Chem. 2008;51(5):1203–13. doi:10.1021/jm700972e.
Napper AD, Hixon J, McDonagh T, Keavey K, Pons JF, Barker J, et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J Med Chem. 2005;48(25):8045–54. doi:10.1021/jm050522v.
Gertz M, Fischer F, Nguyen GT, Lakshminarasimhan M, Schutkowski M, Weyand M, et al. Ex-527 inhibits Sirtuins by exploiting their unique NAD + -dependent deacetylation mechanism. Proc Natl Acad Sci U S A. 2013;110(30):E2772–81. doi:10.1073/pnas.1303628110.
Solomon JM, Pasupuleti R, Xu L, McDonagh T, Curtis R, DiStefano PS, et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol Cell Biol. 2006;26(1):28–38. doi:10.1128/MCB.26.1.28-38.2006.
Sussmuth SD, Haider S, Landwehrmeyer GB, Farmer R, Frost C, Tripepi G, et al. An exploratory double-blind, randomized clinical trial with selisistat, a SirT1 inhibitor, in patients with Huntington’s disease. Br J Clin Pharmacol. 2015;79(3):465–76. doi:10.1111/bcp.12512.
Langsfeld ES, Bodily JM, Laimins LA. The deacetylase sirtuin 1 regulates human papillomavirus replication by modulating histone acetylation and recruitment of DNA damage factors NBS1 and Rad51 to viral genomes. PLoS Pathog. 2015;11(9):e1005181. doi:10.1371/journal.ppat.1005181.
Heltweg B, Gatbonton T, Schuler AD, Posakony J, Li H, Goehle S, et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006;66(8):4368–77. doi:10.1158/0008-5472.CAN-05-3617.
Portmann S, Fahrner R, Lechleiter A, Keogh A, Overney S, Laemmle A, et al. Antitumor effect of SIRT1 inhibition in human HCC tumor models in vitro and in vivo. Mol Cancer Ther. 2013;12(4):499–508. doi:10.1158/1535-7163.MCT-12-0700.
Marshall GM, Liu PY, Gherardi S, Scarlett CJ, Bedalov A, Xu N, et al. SIRT1 promotes N-Myc oncogenesis through a positive feedback loop involving the effects of MKP3 and ERK on N-Myc protein stability. PLoS Genet. 2011;7(6):e1002135. doi:10.1371/journal.pgen.1002135.
Medda F, Russell RJ, Higgins M, McCarthy AR, Campbell J, Slawin AM, et al. Novel cambinol analogs as sirtuin inhibitors: synthesis, biological evaluation, and rationalization of activity. J Med Chem. 2009;52(9):2673–82. doi:10.1021/jm8014298.
Rotili D, Carafa V, Tarantino D, Botta G, Nebbioso A, Altucci L, et al. Simplification of the tetracyclic SIRT1-selective inhibitor MC2141: coumarin- and pyrimidine-based SIRT1/2 inhibitors with different selectivity profile. Bioorg Med Chem. 2011;19(12):3659–68. doi:10.1016/j.bmc.2011.01.025.
Mahajan SS, Scian M, Sripathy S, Posakony J, Lao U, Loe TK, et al. Development of pyrazolone and isoxazol-5-one cambinol analogues as sirtuin inhibitors. J Med Chem. 2014;57(8):3283–94. doi:10.1021/jm4018064.
He X, Nie H, Hong Y, Sheng C, Xia W, Ying W. SIRT2 activity is required for the survival of C6 glioma cells. Biochem Biophys Res Commun. 2012;417(1):468–72. doi:10.1016/j.bbrc.2011.11.141.
Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008;13(5):454–63. doi:10.1016/j.ccr.2008.03.004.
Hirai S, Endo S, Saito R, Hirose M, Ueno T, Suzuki H, et al. Antitumor effects of a sirtuin inhibitor, tenovin-6, against gastric cancer cells via death receptor 5 up-regulation. PLoS ONE. 2014;9(7):e102831. doi:10.1371/journal.pone.0102831.
Yuan H, Wang Z, Li L, Zhang H, Modi H, Horne D, et al. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood. 2012;119(8):1904–14. doi:10.1182/blood-2011-06-361691.
Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J Biol Chem. 2001;276(42):38837–43. doi:10.1074/jbc.M106779200.
Ota H, Tokunaga E, Chang K, Hikasa M, Iijima K, Eto M, et al. Sirt1 inhibitor, sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene. 2006;25(2):176–85. doi:10.1038/sj.onc.1209049.
Kojima K, Ohhashi R, Fujita Y, Hamada N, Akao Y, Nozawa Y, et al. A role for SIRT1 in cell growth and chemoresistance in prostate cancer PC3 and DU145 cells. Biochem Biophys Res Commun. 2008;373(3):423–8. doi:10.1016/j.bbrc.2008.06.045.
Jin KL, Park JY, Noh EJ, Hoe KL, Lee JH, Kim JH, et al. The effect of combined treatment with cisplatin and histone deacetylase inhibitors on HeLa cells. J Gynecol Oncol. 2010;21(4):262–8. doi:10.3802/jgo.2010.21.4.262.
Kozako T, Aikawa A, Shoji T, Fujimoto T, Yoshimitsu M, Shirasawa S, et al. High expression of the longevity gene product SIRT1 and apoptosis induction by sirtinol in adult T-cell leukemia cells. Int J Cancer J Int du Cancer. 2012;131(9):2044–55. doi:10.1002/ijc.27481.
Lara E, Mai A, Calvanese V, Altucci L, Lopez-Nieva P, Martinez-Chantar ML, et al. Salermide, a sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene. 2009;28(6):781–91. doi:10.1038/onc.2008.436.
Liu PY, Xu N, Malyukova A, Scarlett CJ, Sun YT, Zhang XD, et al. The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death Differ. 2013;20(3):503–14. doi:10.1038/cdd.2012.147.
Rotili D, Tarantino D, Nebbioso A, Paolini C, Huidobro C, Lara E, et al. Discovery of salermide-related sirtuin inhibitors: binding mode studies and antiproliferative effects in cancer cells including cancer stem cells. J Med Chem. 2012;55(24):10937–47. doi:10.1021/jm3011614.
Pasco MY, Rotili D, Altucci L, Farina F, Rouleau GA, Mai A, et al. Characterization of sirtuin inhibitors in nematodes expressing a muscular dystrophy protein reveals muscle cell and behavioral protection by specific sirtinol analogues. J Med Chem. 2010;53(3):1407–11. doi:10.1021/jm9013345.
Lancelot J, Caby S, Dubois-Abdesselem F, Vanderstraete M, Trolet J, Oliveira G, et al. Schistosoma mansoni sirtuins: characterization and potential as chemotherapeutic targets. PLoS Negl Trop Dis. 2013;7(9):e2428. doi:10.1371/journal.pntd.0002428.
Rotili D, Tarantino D, Carafa V, Lara E, Meade S, Botta G, et al. Identification of tri- and tetracyclic pyrimidinediones as sirtuin inhibitors. ChemMedChem. 2010;5(5):674–7. doi:10.1002/cmdc.201000030.
Rotili D, Tarantino D, Carafa V, Paolini C, Schemies J, Jung M, et al. Benzodeazaoxaflavins as sirtuin inhibitors with antiproliferative properties in cancer stem cells. J Med Chem. 2012;55(18):8193–7. doi:10.1021/jm301115r.
Zhang Q, Zeng SX, Zhang Y, Zhang Y, Ding D, Ye Q, et al. A small molecule inauhzin inhibits SIRT1 activity and suppresses tumour growth through activation of p53. EMBO Mol Med. 2012;4(4):298–312. doi:10.1002/emmm.201100211.
Disch JS, Evindar G, Chiu CH, Blum CA, Dai H, Jin L, et al. Discovery of thieno[3,2-d]pyrimidine-6-carboxamides as potent inhibitors of SIRT1, SIRT2, and SIRT3. J Med Chem. 2013;56(9):3666–79. doi:10.1021/jm400204k.
Rumpf T, Schiedel M, Karaman B, Roessler C, North BJ, Lehotzky A, et al. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat Commun. 2015;6:6263. doi:10.1038/ncomms7263.
Schiedel M, Rumpf T, Karaman B, Lehotzky A, Olah J, Gerhardt S et al. Aminothiazoles as potent and selective Sirt2 inhibitors: a structure-activity relationship study. J Med Chem. 2016. doi:10.1021/acs.jmedchem.5b01517
Cui H, Kamal Z, Ai T, Xu Y, More SS, Wilson DJ, et al. Discovery of potent and selective sirtuin 2 (SIRT2) inhibitors using a fragment-based approach. J Med Chem. 2014;57(20):8340–57. doi:10.1021/jm500777s.
Friden-Saxin M, Seifert T, Landergren MR, Suuronen T, Lahtela-Kakkonen M, Jarho EM, et al. Synthesis and evaluation of substituted chroman-4-one and chromone derivatives as sirtuin 2-selective inhibitors. J Med Chem. 2012;55(16):7104–13. doi:10.1021/jm3005288.
Seifert T, Malo M, Kokkola T, Engen K, Friden-Saxin M, Wallen EA, et al. Chroman-4-one- and chromone-based sirtuin 2 inhibitors with antiproliferative properties in cancer cells. J Med Chem. 2014;57(23):9870–88. doi:10.1021/jm500930h.
Taylor DM, Balabadra U, Xiang Z, Woodman B, Meade S, Amore A, et al. A brain-permeable small molecule reduces neuronal cholesterol by inhibiting activity of sirtuin 2 deacetylase. ACS Chem Biol. 2011;6(6):540–6. doi:10.1021/cb100376q.
Chopra V, Quinti L, Kim J, Vollor L, Narayanan KL, Edgerly C, et al. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington’s disease mouse models. Cell Rep. 2012;2(6):1492–7. doi:10.1016/j.celrep.2012.11.001.
Chen X, Wales P, Quinti L, Zuo F, Moniot S, Herisson F, et al. The sirtuin-2 inhibitor AK7 is neuroprotective in models of Parkinson’s disease but not amyotrophic lateral sclerosis and cerebral ischemia. PLoS ONE. 2015;10(1):e0116919. doi:10.1371/journal.pone.0116919.
Patel K, Sherrill J, Mrksich M, Scholle MD. Discovery of SIRT3 inhibitors using SAMDI mass spectrometry. J Biomol Screen. 2015;20(7):842–8. doi:10.1177/1087057115588512.
Parenti MD, Grozio A, Bauer I, Galeno L, Damonte P, Millo E, et al. Discovery of novel and selective SIRT6 inhibitors. J Med Chem. 2014;57(11):4796–804. doi:10.1021/jm500487d.
Fatkins DG, Monnot AD, Zheng W. Nepsilon-thioacetyl-lysine: a multi-facet functional probe for enzymatic protein lysine Nepsilon-deacetylation. Bioorg Med Chem Lett. 2006;16(14):3651–6. doi:10.1016/j.bmcl.2006.04.075.
Smith BC, Denu JM. Acetyl-lysine analog peptides as mechanistic probes of protein deacetylases. J Biol Chem. 2007;282(51):37256–65. doi:10.1074/jbc.M707878200.
Smith BC, Denu JM. Mechanism-based inhibition of Sir2 deacetylases by thioacetyl-lysine peptide. Biochemistry. 2007;46(50):14478–86. doi:10.1021/bi7013294.
Fatkins DG, Zheng W. Substituting N(epsilon)-thioacetyl-lysine for N(epsilon)-acetyl-lysine in peptide substrates as a general approach to inhibiting human NAD(+)-dependent protein deacetylases. Int J Mol Sci. 2008;9(1):1–11.
Kiviranta PH, Suuronen T, Wallen EA, Leppanen J, Tervonen J, Kyrylenko S, et al. N(epsilon)-thioacetyl-lysine-containing tri-, tetra-, and pentapeptides as SIRT1 and SIRT2 inhibitors. J Med Chem. 2009;52(7):2153–6. doi:10.1021/jm801401k.
Asaba T, Suzuki T, Ueda R, Tsumoto H, Nakagawa H, Miyata N. Inhibition of human sirtuins by in situ generation of an acetylated lysine-ADP-ribose conjugate. J Am Chem Soc. 2009;131(20):6989–96. doi:10.1021/ja807083y.
Suzuki T, Asaba T, Imai E, Tsumoto H, Nakagawa H, Miyata N. Identification of a cell-active non-peptide sirtuin inhibitor containing N-thioacetyl lysine. Bioorg Med Chem Lett. 2009;19(19):5670–2. doi:10.1016/j.bmcl.2009.08.028.
Huhtiniemi T, Salo HS, Suuronen T, Poso A, Salminen A, Leppanen J, et al. Structure-based design of pseudopeptidic inhibitors for SIRT1 and SIRT2. J Med Chem. 2011;54(19):6456–68. doi:10.1021/jm200590k.
Mellini P, Kokkola T, Suuronen T, Salo HS, Tolvanen L, Mai A, et al. Screen of pseudopeptidic inhibitors of human sirtuins 1-3: two lead compounds with antiproliferative effects in cancer cells. J Med Chem. 2013;56(17):6681–95. doi:10.1021/jm400438k.
Kokkonen P, Rahnasto-Rilla M, Kiviranta PH, Huhtiniemi T, Laitinen T, Poso A, et al. Peptides and pseudopeptides as SIRT6 deacetylation inhibitors. ACS Med Chem Lett. 2012;3(12):969–74. doi:10.1021/ml300139n.
Jamonnak N, Fatkins DG, Wei L, Zheng W. N(epsilon)-methanesulfonyl-lysine as a non-hydrolyzable functional surrogate for N(epsilon)-acetyl-lysine. Org Biomol Chem. 2007;5(6):892–6. doi:10.1039/b617185k.
Huhtiniemi T, Suuronen T, Lahtela-Kakkonen M, Bruijn T, Jaaskelainen S, Poso A, et al. N(epsilon)-modified lysine containing inhibitors for SIRT1 and SIRT2. Bioorg Med Chem. 2010;18(15):5616–25. doi:10.1016/j.bmc.2010.06.035.
Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425(6954):191–6. doi:10.1038/nature01960.
Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430(7000):686–9. doi:10.1038/nature02789.
Valenzano DR, Terzibasi E, Genade T, Cattaneo A, Domenici L, Cellerino A. Resveratrol prolongs lifespan and retards the onset of age-related markers in a short-lived vertebrate. Curr Biol. 2006;16(3):296–300. doi:10.1016/j.cub.2005.12.038.
Rascon B, Hubbard BP, Sinclair DA, Amdam GV. The lifespan extension effects of resveratrol are conserved in the honey bee and may be driven by a mechanism related to caloric restriction. Aging. 2012;4(7):499–508.
Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450(7170):712–6. doi:10.1038/nature06261.
Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444(7117):337–42. doi:10.1038/nature05354.
Minor RK, Baur JA, Gomes AP, Ward TM, Csiszar A, Mercken EM, et al. SRT1720 improves survival and healthspan of obese mice. Sci Rep. 2011;1:70. doi:10.1038/srep00070.
Dai H, Kustigian L, Carney D, Case A, Considine T, Hubbard BP, et al. SIRT1 activation by small molecules: kinetic and biophysical evidence for direct interaction of enzyme and activator. J Biol Chem. 2010;285(43):32695–703. doi:10.1074/jbc.M110.133892.
Hubbard BP, Gomes AP, Dai H, Li J, Case AW, Considine T, et al. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science. 2013;339(6124):1216–9. doi:10.1126/science.1231097.
Sinclair DA, Guarente L. Small-molecule allosteric activators of sirtuins. Annu Rev Pharmacol Toxicol. 2014;54:363–80. doi:10.1146/annurev-pharmtox-010611-134657.
Hubbard BP, Sinclair DA. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci. 2014;35(3):146–54. doi:10.1016/j.tips.2013.12.004.
Bemis JE, Vu CB, Xie R, Nunes JJ, Ng PY, Disch JS, et al. Discovery of oxazolo[4,5-b]pyridines and related heterocyclic analogs as novel SIRT1 activators. Bioorg Med Chem Lett. 2009;19(8):2350–3. doi:10.1016/j.bmcl.2008.11.106.
Vu CB OC, Perni RB, Disch JS, Szczepankiewicz B, Gualtieri G, et al. Inventor solubilized thiazolopyridines. 2009.
Vu CB DJ, Ng PY, Blum CA, Perni RB. Inventor benzimidazoles and related analogs as sirtuin modulators. 2010.
Borra MT, Langer MR, Slama JT, Denu JM. Substrate specificity and kinetic mechanism of the Sir2 family of NAD + -dependent histone/protein deacetylases. Biochemistry. 2004;43(30):9877–87. doi:10.1021/bi049592e.
Kaeberlein M, McDonagh T, Heltweg B, Hixon J, Westman EA, Caldwell SD, et al. Substrate-specific activation of sirtuins by resveratrol. J Biol Chem. 2005;280(17):17038–45. doi:10.1074/jbc.M500655200.
Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem. 2010;285(11):8340–51. doi:10.1074/jbc.M109.088682.
Park SJ, Ahmad F, Philp A, Baar K, Williams T, Luo H, et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell. 2012;148(3):421–33. doi:10.1016/j.cell.2012.01.017.
Lakshminarasimhan M, Rauh D, Schutkowski M, Steegborn C. Sirt1 activation by resveratrol is substrate sequence-selective. Aging. 2013;5(3):151–4.
Dai H, Case AW, Riera TV, Considine T, Lee JE, Hamuro Y, et al. Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat Commun. 2015;6:7645. doi:10.1038/ncomms8645.
Mai A, Valente S, Meade S, Carafa V, Tardugno M, Nebbioso A, et al. Study of 1,4-dihydropyridine structural scaffold: discovery of novel sirtuin activators and inhibitors. J Med Chem. 2009;52(17):5496–504. doi:10.1021/jm9008289.
Spallotta F, Cencioni C, Straino S, Nanni S, Rosati J, Artuso S, et al. A nitric oxide-dependent cross-talk between class I and III histone deacetylases accelerates skin repair. J Biol Chem. 2013;288(16):11004–12. doi:10.1074/jbc.M112.441816.
Valente S, Mellini P, Spallotta F, Carafa V, Nebbioso A, Polletta L et al. 1,4-Dihydropyridines active on the SIRT1/AMPK pathway ameliorate skin repair and mitochondrial function and exhibit inhibition of proliferation in cancer cells. J Med Chem. 2016. doi:10.1021/acs.jmedchem.5b01117
Pillai VB, Samant S, Sundaresan NR, Raghuraman H, Kim G, Bonner MY, et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat Commun. 2015;6:6656. doi:10.1038/ncomms7656.
Acknowledgements
This work was supported by EU: COST-Action: EPICHEMBIO (CM1406); FP7: BLUEPRINT (contract no 282510) and A-PARADDISE (contract no 602080); PRIN-2012, RF-2010-2318330, IIT-Sapienza Project, Sapienza Award Project 2014, AIRC: (a) Fondazione Cariplo TRIDEO Id. 17515 and (b) AIRC-IG (no 17217). We acknowledge C. Fisher for English editing.
Authors’ contributions
VC and DR reviewed the main literature for biology and chemistry. MF, FC, and ES reviewed the biological effects of sirtuins. GSH, EJ, and MLK studied the chemistry and the peptide-related part. AM and LA wrote the manuscript and the general concept of this review. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Carafa, V., Rotili, D., Forgione, M. et al. Sirtuin functions and modulation: from chemistry to the clinic. Clin Epigenet 8, 61 (2016). https://doi.org/10.1186/s13148-016-0224-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-016-0224-3