
Abstract
Background
Intratumor heterogeneity has been demonstrated in several cancer types, following a model of branched evolution. It is unknown to which extent intratumor heterogeneity is applicable to esophageal adenocarcinoma. Therefore the aim of this study was to characterise intratumor heterogeneity in esophageal adenocarcinoma.
Methods
Multiregional targeted sequencing of four commonly altered genes was performed on 19 tumor regions collected from five esophageal adenocarcinomas. Alterations were classified as homogeneous or heterogeneous based on mutational and loss of heterozygosity analysis.
Results
Identical TP53 mutations and homogeneously loss of heterozygosity of the TP53 locus were identified in all separated tumor regions in each of five adenocarcinomas, and in the corresponding Barrett’s esophagus and tumor positive lymph node of one primary tumor. Loss of heterozygosity of the P16 locus was homogeneous among all tumor regions in four adenocarcinomas, and an identical pattern of loss of heterozygosity was present in the Barrett’s esophagus. Loss of heterozygosity of the SMAD4 and APC loci was observed in a heterogeneous pattern.
Conclusions
Known driver alterations, such as TP53 and P16 are homogeneously present within each adenocarcinoma, and therefore occur early during carcinogenesis and subsequently clonally expand throughout the entire tumor. However, loss of heterozygosity of the SMAD4 and APC loci shows a heterogeneous pattern, indicating intratumor heterogeneity of esophageal adenocarcinoma.
Similar content being viewed by others


Background
The incidence of esophageal adenocarcinoma (EAC) has been rising rapidly in Western countries over the last decades [1,2,3]. The major risk factor for EAC is Barrett’s esophagus (BE) [4], a premalignant condition, in which the normal squamous epithelium of the distal esophagus has been replaced by columnar epithelium, including goblet cells. The risk of developing EAC from BE is estimated at 0.12–0.5% per year and follows a multimorphological sequence, in which metaplasia evolves into low-grade dysplasia (LGD), high-grade dysplasia (HGD) and ultimately into EAC [5,6,7].
Previous results from whole-exome/genome sequencing of EACs demonstrated that these tumors bear a broad mutational spectrum; genes frequently altered are e.g. TP53, P16, SMAD4 and APC [8,9,10]. Alterations in the TP53 gene occur in the majority of EAC cases, however, only few other somatic alterations are shared between EACs, representing substantial intertumor heterogeneity. In addition, emerging evidence suggests that even an extensive molecular variation is present within individual tumors, termed intratumor heterogeneity [11,12,13].
With the use of Next-generation sequencing, intratumor heterogeneity has been demonstrated in several cancer types and even a model of branched evolution leading to intratumor heterogeneity has been advocated [12, 14]. This model implicates that a single biopsy may not represent the total mutational burden of a tumor, which can explain the heterogeneous mutational spectrum described in EACs [8]. Conceivably, intratumor heterogeneity may lead to an underestimation of the mutational spectrum of a tumor using a single biopsy procedure, which might clarify the difficulties in finding and validating clinically valuable oncological biomarkers.
To date it is unknown to which extent this model of branched evolution for intratumor heterogeneity is applicable to EAC. Therefore, the aim of this study was to characterize intratumor heterogeneity in EAC, by multiregional targeted sequencing on a total of 19 tumor regions collected from five EAC patients, including adjacent BE in one of them, who underwent primary surgical resection with curative intent. To evaluate intratumor heterogeneity, DNA alterations were classified as homogeneous, i.e. present in all regions of the tumor, or heterogeneous defined as present in some regions or in only one region of the tumor.
Methods
Patients and specimens
Resection specimens of distal esophageal or gastro-esophageal junction adenocarcinomas were obtained from five patients treated between December 2001 and April 2002 at the Department of Surgery, Erasmus MC Cancer Institute, University Medical Center Rotterdam, The Netherlands. All patients underwent a transhiatal esophagectomy, and none of them received neoadjuvant chemo- or radiation therapy, which is part of the current standard treatment of EAC [15].
Tissue samples, DNA isolation and immunohistochemistry
Tissue samples were derived from non-malignant and malignant areas in the fresh resection specimens and used according to the Code of Proper Secondary Use of Human Tissue in the Netherlands, as established by the Dutch Federation of Medical Scientific Societies (http://www.federa.org). In addition the study was approved by The Medical Ethical Committee of the Erasmus MC, University Medical Center, Rotterdam (MEC-2016-067).
From each of the five EACs, multiple, macroscopically separated tumor regions were collected (Fig. 1). In addition, from one resection specimen, samples from an area of premalignant BE (HGD) and from a tumor positive lymph node (TPLN) were available for study. Tumor tissue areas composed of at least 50% neoplastic cells (confirmed by a GI-pathologist) were manually microdissected from 10 to 15 hematoxylin-stained Sects. (4 µm) of formalin-fixed paraffin-embedded tissue blocks. DNA was extracted using proteinase K and 5% Chelex 100 resin. TP53 immunohistochemistry was performed with the mouse monoclonal antibody Do-7 (Dako, Glostrup, Denmark), according to standard protocols.

Macroscopically separated tumor regions of resection specimen
Ion torrent personal genome machine
Ion semiconductor sequencing was performed on the ion torrent personal genome machine (PGM) with a custom-made cancer panel on DNAs extracted from the macroscopically separated tumor regions according to the manufacturer’s protocols. In short, libraries were made using the Ion AmpliSeq Library Preparation Kit. A template was prepared using the Ion OneTouch Template Kit and sequencing was performed with the Ion Sequencing Kit v2.0 on an Ion 316 chip. Data were analysed with the Variant Caller v2.2.3-31149 (Life Technologies, Carlsbad, CA, USA). Variants were called when the position was covered at least 100 times. Sequences of all primers and probes are available on request.
Mutation analysis
The custom-made cancer panel contained the genes: APC, P16, SMAD4 and TP53. Of all genes the total coding regions and the exon–intron boundaries were covered. Nonsynonymous somatic point mutations, insertions and deletions that change the protein amino acid sequence and splice site alterations were selected. In addition, variants present in the ESP6500si or 1000genomes databases in ≥1% were excluded. Variants present in at least 30% of the called reads were considered reliable and were validated by Sanger sequencing according to standard protocols.
Loss of heterozygosity analysis
To demonstrate loss of heterozygosity (LOH) of the genes of interest (APC, P16, SMAD4, TP53) amplicons for single nucleotide polymorphisms (SNPs, selected from dbSNP137) in these loci were added to the custom-made cancer panel. Per locus nine SNPs were selected, all with a minor allele frequency of ≥0.45 and located within the gene (three SNPs) and at positions ~300, ~600 and ~900 kb centromeric and telomeric of the gene. The SNPs were considered heterozygous if the percentage of the variant in the normal DNA was within the range of 40–60%. When at least one SNP was present heterozygous in the normal DNA it was considered informative for LOH analysis. Variants present in the normal DNA in <10% were considered homozygous reference and therefore not informative for LOH analysis, as well as variants present in >90%, which were considered homozygous variant.
For tumor DNAs, of which the normal DNA was considered heterozygous, all SNPs with variants <40 or >60% were denoted as indicative for LOH. A sample was evaluated as having locus LOH when ≥50% of the informative SNPs in that locus demonstrated LOH (variant in tumor DNA <40 or >60%) [16]. In addition, identified tumor suppressor gene mutations also supplied information about possible LOH (loss of the wild type allele) by the relative frequency of the mutant DNA sequence compared to the normal wild type sequence in the tumor samples.
Intratumor heterogeneity analysis
Analysis of intratumor heterogeneity could be determined through mutation and LOH analysis. Homogeneity was signified by an identical mutation or the same pattern of LOH at a given gene identified in different tumor regions of the same tumor, whereas heterogeneity was signified by different mutations or various LOH patterns of a given gene in different regions of the same tumor.
Results
Targeted sequencing with a custom-made cancer panel was performed on DNA isolated from 19 tumor regions derived from the resection specimen of five EAC patients; all male, with a median age at the time of diagnosis of 70 years (range 51–78 years). Two tumors were localised at the gastro-esophageal junction and three in the distal esophagus within the background of BE. Four tumors were moderately differentiated, and one tumor was poorly differentiated. All resection margins were free of tumor cells. Three patients had tumor positive lymph nodes, without distant metastasis. Patient and tumor characteristics are listed in Table 1.
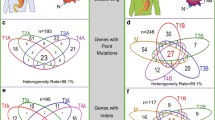
From one resection specimen, a region of BE (HGD) and a TPLN were analysed with the custom-made cancer panel as well. Next-generation sequencing on the PGM and conventional Sanger sequencing revealed 21 mutations in the TP53 gene, no mutations were identified in APC, P16, and SMAD4. Reliable data for the LOH analysis of TP53, P16, SMAD4 and APC were obtained in 92, 92, 81 and 73% of the samples, respectively.
All five EACs showed homogeneous TP53 mutations: in each EAC the same TP53 mutations (nonsynonymous somatic point mutations or splice site alterations) were identified in all investigated tumor regions, all mutations were previous described in esophageal cancer samples in the COSMIC database. In addition, the TP53 mutation found in a primary EAC was also identified in the adjacent BE and TPLN samples. LOH of TP53 occurred homogeneously in all informative regions of the EACs and in the paired EAC and BE samples with identical LOH patterns observed by SNP and/or mutation analysis (Fig. 2). TP53 immunohistochemistry showed homogeneous and strong nuclear expression in all tumor regions and the BE sample of the four patients with TP53 somatic missense mutations (Fig. 3). The tumor cells of the four regions derived from the EAC with a TP53 splice site mutation were all homogeneously negative for TP53 expression (EAC2).

Overview of mutation and LOH analysis per tumor regions of five EACs

TP53 immunohistochemistry on Barrett’s high-grade dysplasia and invasive EAC tissue
LOH of P16 occurred homogeneously in all regions of four informative tumors (EAC1, 2, 4, 5), and an identical LOH pattern of P16 was observed in the paired tumor and BE samples (Fig. 4). EAC1 and EAC3 showed homogeneous LOH of SMAD4 in all tumor regions, two tumors showed different subclones with LOH or without LOH of SMAD4 (EAC2 and 5), while in EAC4 no LOH of SMAD4 was identified. Homogeneous LOH of APC was observed in two tumors (EAC1 and 2), different subclones with and without LOH of APC were observed in EAC3 and 4, whereas one tumor (EAC5) was not informative for the APC locus (Fig. 2).

P16 LOH SNP analysis of normal, Barrett’s high-grade dysplasia and concomitant EAC sample
Discussion
Recent data on Next-generation sequencing of several tumor DNAs were supportive for the model of branched tumor evolution leading to intratumor heterogeneity [11, 13]. This model describes a tumor as a tree structure, with the trunk representing early molecular alterations, which clonally expand and therefore are homogeneously present throughout the entire tumor, reflecting a process involved before and during tumor initiation and early development. The branches of the tree represent later molecular alterations, which as a result are only present in different subclones of the tumor, contributing to intratumor heterogeneity and shaping the genome during tumor maintenance and progression [12, 14]. The extent of this branched evolution model leading to intratumor heterogeneity in EACs is as yet unknown.
Therefore, the aim of the current study was to characterize intratumor heterogeneity in EACs, by performing multi-region, targeted sequencing on 19 tumor regions derived from five surgical resected EACs. Targeted sequencing of commonly altered genes; TP53, P16, SMAD4 and APC was performed. This analysis revealed a clonal origin of TP53 alterations: all five EACs where homogeneous with regard to TP53 mutations and LOH of the TP53 locus, in addition the same mutation and pattern of LOH in TP53 was observed in a paired TPLN of one EAC. These results indicate that TP53 mutation and LOH of the TP53 locus are relatively early events in EAC tumorigenesis and clonally expand throughout the entire tumor. In concordance with this is the finding of the same TP53 mutation, identical pattern of LOH, and comparable strong nuclear TP53 expression in the paired primary tumor and BE case.
Mutations in the P16 gene were not observed in this study, however LOH of the P16 locus was identified in all EACs. Four EACs had a homogeneous P16 locus LOH pattern, while one EAC was heterogeneous for P16 LOH. In addition, an identical pattern of P16 locus LOH was present in the paired primary EAC and BE case, suggesting that LOH of P16 is an early alteration, clonally expanding throughout the tumor. No mutations were identified in SMAD4 and APC, but LOH of these genes was observed in a heterogeneous pattern within the EACs. Some EACs showed homogeneous LOH of SMAD4 and/or APC, one EAC showed no LOH of SMAD4 at all, while the remaining EACs showed different subclones: some with LOH of SMAD4 and/or APC and others without LOH of these loci. In addition, no LOH of SMAD4 and APC was found in the BE sample, indicating a late occurrence of these alterations, reflecting intratumor heterogeneity of EACs.
Taken together, the results of the current study suggest that both homogeneous and heterogeneous intratumoral molecular alterations are present in EACs. A homogeneously present TP53 mutation and LOH of the TP53 locus as well as LOH of the P16 locus in the primary tumor were also found in the adjacent BE sample, indicating that the earliest molecular alterations can already be present in the premalignant lesion (BE) and from there on clonally expand throughout the entire tumor. Temporarily, no multiregion sequencing of EACs was performed before. Alterations in TP53 were described previously in the sequential of BE (HGD) and EAC, and in addition were found to be present in a major clone, which also indicates clonal expansion of TP53 as an early alteration [17].
The presence of alterations in BE is in accordance with previous studies, showing that multiple BE crypts contain different clones with alterations competing with each other [18, 19], of which one progenitor clone with a selective growth advantage will expand clonally and will create a field in which other (pre)malignant alterations might arise [19]. Several studies on P16 have reported clonal expansion of P16 alterations in all regions of BE segments, and it has been suggested that expansion of TP53 alterations occurs only in a background of P16 altered clones [20]. However, others described progenitor clones containing TP53 alterations alone or in combination with P16 alterations present at many levels of BE segments [21]. Leedham et al. [18] concluded that BE was genetically heterogeneous, however they observed identical TP53 mutations in multiple BE crypts, indicating widespread and far-reaching clonal expansion as a consequence of the strong selective advantage that absence of TP53 function supposedly provide. Recently, a study on cytochrome c oxidase (CCO) deficient cells in BE confirmed the concept of clonal expansion of BE, probably by fission of BE glands [22].
Conceivably, intratumor heterogeneity can cause tumor sample bias using single biopsy approaches, since it may underestimate the mutation spectrum of a tumor. This could contribute to difficulties in identifying and validating biomarkers, which are desirable to identify BE patients with a high risk for neoplastic progression. Furthermore, intratumor heterogeneity may contribute to therapy resistance: if the actionable target of treatment is only present in a subclone of the tumor, than targeting this genetic alteration may not have an impact on the entire tumor. This concept might explain the diversity in responders and non-responders after neoadjuvant chemoradiotherapy in EACs [23].
Conclusions
Recent studies on EAC reveal extensive intertumor heterogeneity concerning molecular alterations with different studies showing different recurrently mutated genes. Therefore, the genes found frequently mutated in several studies were investigated in the current study [8,9,10, 17]. Even though the sample size is small, and targeted sequencing of only commonly mutated genes was performed, the current study provides evidence that although intratumor heterogeneity is present in EACs, known driver alterations in TP53 and P16 were homogeneously present in all five primary EACs, indicating clonal cellular expansion. Studies with larger cohorts and extensive genome sequencing are needed to fully characterize intratumor heterogeneity in EACs and in addition, to understand the impact of intratumor heterogeneity on current clinical outcome of both BE and EAC patients.
References
Bosetti C, Levi F, Ferlay J, Garavello W, Lucchini F, Bertuccio P, Negri E, La Vecchia C. Trends in oesophageal cancer incidence and mortality in Europe. Int J Cancer. 2008;122(5):1118–29.
Brown LM, Devesa SS, Chow WH. Incidence of adenocarcinoma of the esophagus among white Americans by sex, stage, and age. J Natl Cancer Inst. 2008;100(16):1184–7.
Dikken JL, Lemmens VE, Wouters MW, Wijnhoven BP, Siersema PD, Nieuwenhuijzen GA, van Sandick JW, Cats A, Verheij M, Coebergh JW, et al. Increased incidence and survival for oesophageal cancer but not for gastric cardia cancer in the Netherlands. Eur J Cancer. 2012;48(11):1624–32.
Cameron AJ. Epidemiology of columnar-lined esophagus and adenocarcinoma. Gastroenterol Clin North Am. 1997;26(3):487–94.
Bhat S, Coleman HG, Yousef F, Johnston BT, McManus DT, Gavin AT, Murray LJ. Risk of malignant progression in Barrett’s esophagus patients: results from a large population-based study. J Natl Cancer Inst. 2011;103(13):1049–57.
Hvid-Jensen F, Pedersen L, Drewes AM, Sorensen HT, Funch-Jensen P. Incidence of adenocarcinoma among patients with Barrett’s esophagus. N Engl J Med. 2011;365(15):1375–83.
Jankowski JA, Wright NA, Meltzer SJ, Triadafilopoulos G, Geboes K, Casson AG, Kerr D, Young LS. Molecular evolution of the metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am J Pathol. 1999;154(4):965–73.
Dulak AM, Stojanov P, Peng S, Lawrence MS, Fox C, Stewart C, Bandla S, Imamura Y, Schumacher SE, Shefler E, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013;45(5):478–86.
Streppel MM, Lata S, DelaBastide M, Montgomery EA, Wang JS, Canto MI, Macgregor-Das AM, Pai S, Morsink FH, Offerhaus GJ, et al. Next-generation sequencing of endoscopic biopsies identifies ARID1A as a tumor-suppressor gene in Barrett’s esophagus. Oncogene. 2014;33(3):347–57.
Agrawal N, Jiao Y, Bettegowda C, Hutfless SM, Wang Y, David S, Cheng Y, Twaddell WS, Latt NL, Shin EJ, et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2012;2(10):899–905.
de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, Jamal-Hanjani M, Shafi S, Murugaesu N, Rowan AJ, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346(6206):251–6.
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–92.
Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, Seth S, Chow CW, Cao Y, Gumbs C, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014;346(6206):256–9.
Yap TA, Gerlinger M, Futreal PA, Pusztai L, Swanton C. Intratumor heterogeneity: seeing the wood for the trees. Sci Transl Med. 2012;4(127):127ps110.
van Hagen P, Hulshof MC, van Lanschot JJ, Steyerberg EW, van Berge Henegouwen MI, Wijnhoven BP, Richel DJ, Nieuwenhuijzen GA, Hospers GA, Bonenkamp JJ, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Engl J Med. 2012;366(22):2074–84.
Dubbink HJ, Atmodimedjo PN, van Marion R, Krol NM, Riegman PH, Kros JM, van den Bent MJ, Dinjens WN. Diagnostic detection of allelic losses and imbalances by next generation sequencing: 1p/19q co-deletion analysis of gliomas. J Mol Diagn. 2016;18(5):775–86.
Weaver JM, Ross-Innes CS, Shannon N, Lynch AG, Forshew T, Barbera M, Murtaza M, Ong CA, Lao-Sirieix P, Dunning MJ, et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat Genet. 2014;46(8):837–43.
Leedham SJ, Preston SL, McDonald SA, Elia G, Bhandari P, Poller D, Harrison R, Novelli MR, Jankowski JA, Wright NA. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s oesophagus. Gut. 2008;57(8):1041–8.
Wong DJ, Paulson TG, Prevo LJ, Galipeau PC, Longton G, Blount PL, Reid BJ. p16(INK4a) lesions are common, early abnormalities that undergo clonal expansion in Barrett’s metaplastic epithelium. Cancer Res. 2001;61(22):8284–9.
Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Reid BJ. Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett’s esophagus. Cancer Res. 2004;64(10):3414–27.
Barrett MT, Sanchez CA, Prevo LJ, Wong DJ, Galipeau PC, Paulson TG, Rabinovitch PS, Reid BJ. Evolution of neoplastic cell lineages in Barrett oesophagus. Nat Genet. 1999;22(1):106–9.
Nicholson AM, Graham TA, Simpson A, Humphries A, Burch N, Rodriguez-Justo M, Novelli M, Harrison R, Wright NA, McDonald SA, et al. Barrett’s metaplasia glands are clonal, contain multiple stem cells and share a common squamous progenitor. Gut. 2012;61(10):1380–9.
Reynolds JV, Muldoon C, Hollywood D, Ravi N, Rowley S, O’Byrne K, Kennedy J, Murphy TJ. Long-term outcomes following neoadjuvant chemoradiotherapy for esophageal cancer. Ann Surg. 2007;245(5):707–16.
Authors’ contributions
AM Conception and design, acquisition of data, analysis and interpretation of data, drafting the manuscript. R Conception and design, acquisition of data, analysis and interpretation of data, have given final approval of the version to be published. LB Conception and design, acquisition of data, analysis and interpretation of data, involved in drafting the manuscript, have given final approval of the version to be published. K acquisition of data, revising the manuscript critically for important intellectual content, have given final approval of the version to be published. MC revising the manuscript critically for important intellectual content, have given final approval of the version to be published. HW revising the manuscript critically for important intellectual content, have given final approval of the version to be published. JJ revising the manuscript critically for important intellectual content, have given final approval of the version to be published. BP Conception and design, involved drafting the manuscript, have given final approval of the version to be published. WN Conception and design, analysis and interpretation of data, involved in drafting the manuscript, have given final approval of the version to be published. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank Gerard Gathier and Ludo Uytdewilligen from the Laboratory for Molecular Diagnostics, Department of Pathology for preparing the tissues and performing sequencing.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The data and material supporting the conclusions of this article are included within the article and its additional files.
Ethics approval and consent to participate
Tissue samples were derived from resection specimens of deceased patients, and therefore written informed consent could not be obtained. However, tissue samples were used according to the Code of Proper Secondary Use of Human Tissue in the Netherlands, as established by the Dutch Federation of Medical Scientific Societies (http://www.federa.org). In addition the study was approved by The Medical Ethical Committee of the Erasmus MC, University Medical Center, Rotterdam (MEC-2016-067).
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
van Nistelrooij, A.M.J., van Marion, R., Koppert, L.B. et al. Molecular clonality analysis of esophageal adenocarcinoma by multiregion sequencing of tumor samples. BMC Res Notes 10, 144 (2017). https://doi.org/10.1186/s13104-017-2456-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13104-017-2456-5