
Abstract
Background
To determine the effect of riociguat, an oral, selective soluble guanylate cyclase stimulator, on the net digital ulcer (DU) burden in systemic sclerosis (SSc).
Methods
Participants with SSc-related active or painful indeterminate DUs were recruited in a multicenter, double-blind, randomized, placebo-controlled, proof-of-concept trial. Eligible participants were required to have at least one visible, active ischemic DU or painful indeterminate DU at screening, located at or distal to the proximal interphalangeal joint and that developed or worsened within 8 weeks prior to screening. Participants were randomized 1:1 to placebo or riociguat in individualized doses (maximum of 2.5 mg three times daily) during an 8-week titration period, followed by an 8-week stable dosing period. This was followed by an optional 16-week open-label extension phase for participants with active DU/reoccurrence of DUs within 1 month of the end of the main treatment phase. The primary endpoint was the change from baseline to week 16 in net ulcer burden (NUB), analyzed using ANCOVA. Other endpoints included plasma biomarkers and proportion of participants with treatment-emergent adverse events (AEs).
Results
Seventeen participants (eight placebo, nine riociguat) were randomized at five centers. Six participants in each group transitioned to the open-label extension. Baseline characteristics were comparable between the treatment groups, except participants randomized to placebo were older and had longer disease duration (p < 0.05). At baseline, the mean (SD) NUB was 2.5 (2.0) in the placebo and 2.4 (1.4) in the riociguat. No significant treatment difference was observed in the change from baseline to 16 weeks in NUB (adjusted mean treatment difference − 0.24, 95% CI (− 1.46, 0.99), p = 0.70). Four participants experienced five serious AE (four in riociguat and one in placebo); none was considered related to study medication. Statistically significant elevation of cGMP was observed at 16 weeks in the riociguat group (p = 0.05); no other biomarkers showed significant changes. In the open-label extension, participants in the riociguat-riociguat arm had complete healing of their DUs.
Conclusion
In participants with SSc-DU, treatment with riociguat did not reduce the number of DU net burden compared with placebo at 16 weeks. Open-label extension suggests that longer duration is needed to promote DU healing, which needs to be confirmed in a new trial.
Trial registration
ClinicalTrials.gov, NCT02915835. Registered on September 27, 2016.
Similar content being viewed by others

Background
Systemic sclerosis (SSc) is an autoimmune disorder featuring chronic, fibrosing, autoimmune responses characterized by small vessel vasculopathy, autoantibody production, and fibroblast dysfunction leading to increased deposition of extracellular matrix [1]. Raynaud’s phenomenon (RP) is an almost universal manifestation of SSc, with 95% of all patients being affected, resulting in digital ulcers (DUs) in approximately 30% of the patients each year. DUs are associated with substantial morbidity (reduced quality of life, pain, disability, and disfigurement) that can escalate to gangrene and amputation in approximately 15% of patients [2, 3]. There are no drugs approved in the USA for the treatment of DUs. Treatments that have shown potential include calcium channel blockers, prostacyclin analogs, and endothelin receptor antagonists. Bosentan, a dual endothelin receptor antagonist, is approved in Europe to reduce the number of new DUs in patients with SSc. Trials and case series show beneficial efficacy of phosphodiesterase 5 (PDE5) inhibitors in healing of SSc-DUs, and this finding is supported by a meta-analysis [4].
Riociguat is the first in class of a new group of compounds, soluble guanylate cyclase (sGC) stimulators. Riociguat directly stimulates sGC, thereby increasing the levels of the signaling molecule cGMP. The cGMP molecule plays a pivotal role in regulating cellular processes, such as vascular tone, proliferation, fibrosis, and inflammation. Riociguat has a dual mode of action, directly stimulating sGC independent of nitric oxide (NO) and increasing the sensitivity of sGC to NO [5, 6]. Riociguat is approved for the treatment of two forms of pulmonary hypertension, pulmonary arterial hypertension (PAH), and chronic thromboembolic pulmonary hypertension [7,8,9]. In pre-clinical studies, riociguat has been shown to have vasodilatory, anti-proliferative, vascular remodeling, anti-fibrotic, and anti-inflammatory properties [10,11,12,13,14,15]. A recent single-dose, crossover trial showed that riociguat was well tolerated in patients with RP and resulted in improved digital blood flow in some patient subsets, with high inter-individual variability [16]. In an exploratory analysis of a phase 2 trial of riociguat in patients with early diffuse cutaneous SSc, there was a numerical tendency toward a reduction of RP symptoms and attack frequency with riociguat treatment compared with placebo [17].
The present proof-of-concept trial was designed to assess the efficacy and safety of 16 weeks of treatment with riociguat in a randomized, placebo-controlled clinical trial in patients with SSc-associated DUs followed by an optional open-label extension for additional 16 weeks.
Methods
Study design
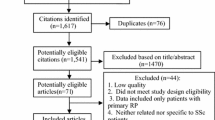
This was an investigator-initiated, multicenter, double-blind, randomized, placebo-controlled, parallel group, proof-of-concept study comprising a 16-week treatment period (8-week titration, 8-week maintenance) followed by 16-week open-label extension phase for participants with active digital ulcer or reoccurrence of DUs within 1 month of the end of the main treatment phase (ClinicalTrials.gov Identifier: NCT02915835). Participants without an ischemic active or painful indeterminate DU after completion of the treatment period had a safety follow-up visit 4-weeks post-treatment. The study was conducted at five scleroderma centers in the USA. The Sponsor (Dinesh Khanna, MD) received an IND exemption from the Food and Drug Administration. Each site’s institutional review board or ethics committee approved the protocol before the study commenced. The study was conducted in accordance with the principles of the Declaration of Helsinki. After providing written informed consent, participants entered a screening phase (lasting up to 2 weeks), where their eligibility was evaluated (Fig. 1). Participants completed a diary detailing the number and duration of Raynaud’s attacks per day for a period of at least seven consecutive days before the baseline visit.

Consort diagram
Randomization and masking
Eligible participants were randomized in a 1:1 ratio to either riociguat or matching placebo (provided by the Bayer). The Data Coordinating Center (DCC) at the University of Michigan prepared the randomization schedule, using permuted block randomization with size 2 for the first block and then the random block sizes of 2 and 4 thereafter (block sizes were known only by the DCC). A secure web-based randomization and drug dispensing application were built by the DCC that was used by coordinators to obtain the randomization number and medication bottle numbers. This information was printed for the subject binder and used to prepare an investigator-signed prescription for the site pharmacist. The study staff (including the research pharmacists and assessors of DU) and participants were blinded to the treatment assigned.
In the titration phase of 8 weeks, participants started at a dose of 1.0 mg three times a day (TID). The individual study medication dose for the next titration step was determined every 2 weeks according to the patient’s well-being and the peripheral systolic blood pressure measured at trough before intake of the morning dose according to the individual dose titration scheme (Fig. 2). The dose was increased by 0.5 mg increments no sooner than 2 weeks (± 4 days) apart to 1.5 mg, 2 mg, and 2.5 mg TID, resulting in a potential maximum total daily dose of 7.5 mg (2.5 mg TID). Participants were maintained on a lower dose if higher doses were not tolerated (minimum dosage of 0.5 mg TID, total daily dose 1.5 mg). While it was possible for a participant to be up-titrated and then down-titrated during this phase, once a participant had been down-titrated, they remained at that dose, and dose escalation was not implemented again. The established individual dose was then taken as the “optimal individual dose” to be administered for the remaining duration of study. To maintain the blinding of the treatment arms, participants randomized to the placebo group underwent sham titration from visit 1 onwards during the dose-titration period.

Study design
At week 16, all participants who agreed to continue in the open-label extension were assigned to treatment with riociguat. During the first 8 weeks of the open-label extension phase, participants previously on placebo were up-titrated on riociguat as per the individual titration algorithm described in the double-blind treatment phase. Participants randomized to riociguat in the double-blind study treatment phase also underwent a titration phase in the open-label extension.
Patient selection
Eligible patients were ≥ 18 years old with a diagnosis of SSc according to the American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) 2013 classification criteria (total score of ≥ 9) [18]. Participants were required to have at least one visible, active ischemic DU or painful indeterminate DU at screening, located at or distal to the proximal interphalangeal joint, and that developed or worsened within 8 weeks prior to screening. An active DU was defined as a full-thickness skin lesion, > 3 mm in maximal diameter, with loss of epithelization, epidermis, and dermis. An indeterminate ulcer was defined as one where denudation could not be visualized and there were no other clinical features of activity. We excluded DUs due to calcinosis (based on baseline hand X-ray in every participant), paronychia, and osteomyelitis. We also excluded fissures, pitting scars, hyperkeratotic lesions, and DUs over the metacarpophalangeal joints (MCPs) or elbows. Photographs of the cardinal DU were taken and confirmed to meet the study inclusion after the review of the photograph by DK. We provided a written standardized wound care for each participant. Females of reproductive potential (FRP) were required to have a negative urine pregnancy test. During treatment, FRP were required to obtain monthly urine pregnancy tests during treatment and 1 month after treatment discontinuation. Oral corticosteroids (≤ 10 mg/day of prednisone or equivalent), non-steroidal anti-inflammatory drugs, angiotensin receptor blockers, angiotensin-converting enzyme inhibitors, and calcium channel blockers were permitted if the participant was on a stable dose for ≥ 2 weeks prior to and including the baseline visit. Participants with sitting systolic blood pressure < 95 mmHg, sitting heart rate < 50 beats/min, left ventricular ejection fraction < 40%, or anemia with hemoglobin < 9.0 g/dl, PAH requiring pharmacologic therapy, significant pulmonary disease [FVC ≤ 50% of predicted, or DLCO (uncorrected for hemoglobin) ≤ 40% of predicted done as part of clinical care], active state of hemoptysis or pulmonary hemorrhage, or any history of bronchial artery embolization or massive hemoptysis within 3 months prior to screening were excluded in the trial. We excluded participants with concomitant use of nitrates or NO donors (such as amyl nitrate) in any form, PDE5 inhibitors (such as sildenafil, tadalafil, or vardenafil due to the risk of hypotension and both target the nitric oxide pathway), and endothelin receptor antagonists. If the participant was on PDE5 inhibitors, a wash out of 3 days was required for sildenafil and 7 days for tadalafil or vardenafil prior to the baseline visit. Patients who were actively smoking at the time of consent were excluded due to the impact of nicotine on pharmacokinetics of riociguat; a quit date of 2 weeks prior to screening was acceptable. Complete inclusion/exclusion criteria are provided in Additional file 1.
Study outcome measures
The primary efficacy endpoint was the change from baseline to week 16 (end of study treatment phase) in net ulcer burden (NUB). NUB is defined as the total number of active and painful indeterminate DUs at an assessment, and the change from baseline in NUB reflects both new DUs as well as healed DUs. For example, if a participant had two active DUs and one painful indeterminate DU at baseline (NUB = 3) and at week 4 had one of the active DUs healed, no change in the other DUs, and development of a two new active DUs (NUB = 4), the change would be one. NUB captures the overall impact of DUs on hand function and quality of life and has been used in a previous trial [2]. Other pre-specified secondary efficacy endpoints included the proportion of participants with the following at week 16: healing of the cardinal DU (healing was defined by re-epithelialization with loss of pain and exudate); healing of all baseline DUs; no DUs; development of new active and indeterminate DUs; development or healing of ulcers over DIP, PIP, MCPs, and elbows; time to healing of the cardinal DU and all baseline DUs; development of new (active or indeterminate) DUs; improvement of RP based on Raynaud’s Condition Score (RCS); number and duration of Raynaud’s attacks per day; patient and physician assessment of RP; symptoms during RP attack (pain, numbness, and tingling); patient’s and physician’s global assessment on a Likert scale; health-related quality of life as measured using Patient-Reported Outcomes Measurement Information System (PROMIS)-29; physical function [as assessed by Health Assessment Questionnaire Disability Index (HAQ-DI); and Hand Disability in Systemic Sclerosis-DU (HDISS-DU)]; visual analog scales from Scleroderma Health Assessment Questionnaire (SHAQ) assessing burden of digital ulcers, Raynaud’s disease, gastrointestinal involvement, breathing, and overall disease; and documentation of digital ischemia requiring intravenous prostacyclin or digital gangrene or amputation.
Biomarker measurement
To examine the effect of riociguat on biomarkers, patient plasma was collected at baseline and week 16. Plasma from age- and sex-matched healthy controls was also obtained. The biomarkers were measured by enzyme-linked immunosorbent assays (ELISA) and included total vascular endothelial growth factor (VEGF, R&D Systems), tissue plasminogen activator (tPA, Abcam), soluble E-selectin (sE-selectin, R&D Systems), basic fibroblast growth factor (bFGF, R&D Systems), vascular cell adhesion molecule 1 (VCAM-1, RayBiotech), soluble intracellular adhesion molecule 1 (sICAM-1, RayBiotech), N-terminal propeptide of type I collagen (PINP, MyBioSource), matrix metalloproteinase 12 (MMP12, RayBiotech), CXCL4 (R&D Systems), cyclic guanylyl cyclase (cGMP, Cayman Chemical), endostatin (R&D Systems), and soluble fms-like tyrosine kinase 1 (sFLT1, R&D Systems).
Sample size
The planned sample size of 20 participants was based primarily on practical considerations, not on power to achieve a pre-determined treatment difference. The goal of this pilot study was to obtain preliminary estimates of the magnitude of treatment effects of key efficacy and safety parameters. There are no published data on the minimal clinically important difference (MCID) for the change from baseline in NUB at week 16 (our primary efficacy endpoint), and our study did not seek to establish the MCID. Rather, we designed this pilot study with a placebo control arm and randomization to reduce bias in the estimation of NUB, so that we could also obtain a preliminary estimate of treatment differences. As expected with a pilot study, only large treatment differences can be detected. With the proposed sample of 10 riociguat and 10 placebo participants, we calculated the effect size (mean treatment difference divided by standard deviation) for the primary efficacy endpoint to be 1.253 with 80% power and a two-sided type I error of 5% based on a two-sample t test. If a statistically significant would be observed in our small study, it would need to be replicated in a larger confirmatory study.
Statistical analysis
Continuous variables were summarized using means, standard deviations (SD), median, interquartile range (IQR), and range, and qualitative variables were summarized using counts and percentages. Mean (SD) is reported, unless otherwise noted. The primary and secondary efficacy endpoints were analyzed using the modified intention-to-treat population (MITT), defined as all participants randomized, receiving at least one dose of treatment, and having at least one post-baseline efficacy assessment. As a sensitivity analysis, the primary endpoint was also analyzed using the per-protocol set, defined as the MITT population who did not have a major protocol violation. For the primary analysis, changes in NUB were compared in the two treatment groups using an ANCOVA model, with terms for treatment group and baseline NUB value. Distributional assumptions were assessed. Analysis for secondary outcome measures that are continuous was performed using a similar approach as that for the primary endpoint. For analyses of discrete secondary outcomes measures, we used Fisher’s exact tests. Poisson regression was used for outcome measures that were counts (e.g., number of AEs) and log-rank tests, and Kaplan-Meier plots were used for time-to-event outcomes. Plasma biomarker changes from baseline (week 0) to week 16 were analyzed using the ANCOVA model. Safety analyses were performed on the safety analysis set which included all participants who were randomized and received at least one dose of the study drug. Statistical tests were conducted at the 0.05 significance level (with no adjustments for multiplicity) using two-tailed tests. Statistical analyses were performed using SAS version 9 or higher. Further details on the statistical analysis can be found in Additional file 2.
Results
Participant disposition and baseline characteristics
Twenty-five participants were screened across 5 centers in the USA between January 2017 and May 2018. Seventeen participants were randomized to either placebo (n = 8) or riociguat (n = 9), of which all 17 (88%) participants formed the MITT and safety analysis sets (Fig. 2), and the trial was stopped in May 2018 due to warm weather and poor recruitment and since this was a proof-of-concept study. Fifteen participants were included in the per-protocol analysis set (8 in placebo and 7 in riociguat). One participant withdrew in each group. One riociguat participant was withdrawn by the investigator due to worsening DU and RP, and the placebo participant completed treatment but did not return for the safety follow-up at week 20. Mean compliance with study drug in the treatment phase was 92%, 96% with placebo, and 88% with riociguat. Six subjects in each group progressed to the open-label extension phase with a 100% compliance till the end of the phase.
Baseline demographics and clinical characteristics were largely similar between the two groups, but the participants in the placebo group had longer duration (in years) of SSc diagnosis (mean [SD] 15.0 [8.2] years vs 6.2 [5.8] years) and of non-Raynaud’s symptoms (17.5 [11.2] years vs 7.1 [6.0] years). (Table 1). At baseline, the mean [SD] NUB was 2.5 (2.0) in the placebo group and 2.4 (1.4) in the riociguat group. Participants randomized to riociguat had numerically worse RP—higher RCS, more frequent and longer Raynaud’s attacks, more intense symptoms associated with RP (pain, numbness, and tingling), and higher S-HAQ scores for DUs indicating increased interferences of DUs with daily activities.
Dosing and exposure
The median duration of exposure to study drug was 112 days in each treatment group. At the end of the 8-week titration phase, all eight participants in the placebo group reached the 2.5 mg TID dosing level whereas in the riociguat group, three reached 1.5 mg TID, one reached 2.0 mg TID, and four reached the 2.5 mg TID dosing levels.
Primary efficacy endpoint
There was no statistically significant difference between riociguat and placebo in the change in NUB (Fig. 3). The least square (LS) mean change from baseline to 16 weeks in NUB was − 1.22 in the riociguat group and − 0.98 in the placebo group (negative score denotes improvement, treatment difference − 0.24, 95% CI − 1.46 to 0.99; p = 0.70; Table 2). Sensitivity analyses (using the per-protocol analysis set or controlling for age in the MITT analysis set) also showed statistically non-significant treatment differences (LS mean treatment difference − 0.08, 95% CI − 1.62 to 1.46; p = 0.92 in ANCOVA adjusting for baseline NUB and age).

Mean trend over time: change in net ulcer burden. Digital ulcer net burden is defined as the total number of “active” and indeterminate digital ulcers at an assessment. LS, least squares; SE, standard error; CI, confidence interval. Estimates are from an ANCOVA model with terms for the treatment group and baseline digital net ulcer burden. Modified intent-to-treat population is defined as all participants randomized, receiving at least one dose of treatment, and having at least one post-baseline efficacy assessment
Secondary endpoints
There were no statistically significant treatment differences in secondary efficacy endpoints, except for the eating component of HAQ-DI (Table 2). There were no statistically significant differences between the two treatments in RCS or frequency of RP attacks, patient self-assessments, PROMIS-29 measures, and overall HAQ-DI score. However, we noted greater numerical improvements in riociguat, relative to placebo, in some outcome measures but were not significant (Table 2). Participants in the riociguat group reported decreased pain due to RP (LS mean treatment difference 6.71, 95% CI − 14.01 to 27.43; p = 0.49) and tingling due to RP (LS mean treatment difference 8.67, 95% CI − 13.75 to 31.09; p = 0.41) during RP attacks and decreased duration of RP attacks (LS mean treatment difference − 195.1 min, 95% CI − 683.7 to 293.5; p = 0.40) compared to the placebo group (both pain and tingling due to RP are measured on a 0–100 scale). In both treatment groups, three (38%) participants underwent healing of all baseline DUs by week 16, with a median time to healing of 16 weeks (placebo, 1.15 riociguat; p = 0.35). Healing of the cardinal DU by week 16 was seen in 6 (75%) placebo and four (50%) riociguat participants. The median time to healing of the cardinal DU was 16.0 weeks (IQR 0.14) in the placebo group and 16.6 weeks (IQR 1.15) in the riociguat group (p = 0.56). The percentage of participants without any DUs at the end of the study period was two (25%) in each of the treatment groups. Four (50%) and three (38%) of placebo and riociguat participants, respectively, developed new ulcers of the 16 weeks of treatment (p = 1.0).
Biomarker data
There were statistical differences in the baseline values for the biomarkers between healthy controls and all patients for cGMP, sE-selection, and sICAM1 (p < 0.05, Additional file 3). No statistical differences were observed in patients in the placebo and riociguat arms at baseline except for MMP12; patients in the riociguat group had significantly higher MMP12 levels compared to the placebo group (p < 0.05, Additional file 3). Our ANCOVA analysis revealed that after 16 weeks of riociguat treatment, there were no significant changes in the biomarkers measured, except for cGMP, which was significantly elevated in the riociguat group, confirming target engagement (p = 0.046, Table 3).
Safety and tolerability
Four serious AEs (SAEs) in 3 (14%) riociguat participants were reported: non-Hodgkin lymphoma, non-ST elevation myocardial infarction, digital ischemia, and worsening digital ulcer that required hospitalization for intravenous prostacyclin. One (8%) placebo participant experienced an SAE: persistent digital ischemia in a toe that required hospitalization for intravenous prostacyclin. All SAEs were considered not related to the study treatment. There were no deaths during the study. Thirteen adverse events (AEs) were reported in 8 (100%) participants in the placebo group and 21 were reported in 9 (100%) participants in the riociguat group (Table 4). Most AEs were reported as mild or moderate according to the Common Terminology Criteria for Adverse Events 5.0 (CTCAE 5.0) severity grading system: 13 (100%) in the placebo group and 77% in the riociguat group. There was no osteomyelitis or AEs of special interest (clinically significant hypotension or hemoptysis).
Open-label extension
There was an improvement in the NUB in both placebo-riociguat and riociguat-riociguat groups from baseline and from week 16 of the treatment phase (Fig. 3). In the riociguat-riociguat group, all the DUs healed by week 16 of the open-label extension. Numerical improvements were noted in almost all the secondary efficacy endpoints by week 16 of the open-label extension in both groups but numerically favor in the riociguat-riociguat group (Table 5). Six SAEs were reported in total. In the placebo-riociguat arm, SAEs included the following: acute ileus (participant #1), omental adhesion of lower abdomen, transient ischemic attack, aspiration pneumonia, and deep vein thrombosis (all in participant #2), and acute respiratory failure secondary to pneumonia (participant #3). In the riociguat-riociguat arm, SAEs included the following: pulmonary embolism and acute right rib fractures with hemopneumothorax (both in participant #1). All SAEs were considered not related to the study treatment. There were no deaths during the open-label extension phase study. Thirty-five AEs were reported in 6 (100%) participants in the placebo-riociguat group and 19 were reported in 5 (83%) participants in the riociguat-riociguat group (Table 4). Most AEs were reported as mild or moderate according to the CTCAE 5.0 severity grading system.
Discussion
The proof-of-concept trial was designed to evaluate the effect of riociguat on NUB in patients with SSc-related DUs. We did not find any statistical or clinically meaningful differences in the NUB and other secondary outcome measures, but we showed target engagement, as exemplified by an increase in plasma cGMP in the riociguat group. With a longer duration of treatment with riociguat, complete healing of DUs was observed as noted in the open-label extension phase. The safety profile of riociguat was consistent with that observed previously in studies of patients with PAH, with no new safety events identified [9].
Few therapies are available for DUs in patients with SSc. In the recently published EULAR guidelines, intravenous iloprost and oral sildenafil are recommended for the treatment of SSc-related DU, and oral bosentan is recommended for the prevention of new DUs, especially in patients with multiple digital ulcers despite the use of CCBs, PDE5 inhibitors, or iloprost therapy [19]. In the USA, iloprost is currently unavailable, and neither sildenafil nor bosentan is approved by the Food and Drug Administration for the treatment of DUs. Hence, there is a clear need for therapeutic agents for the management of DUs.
We designed the pilot trial to explore the unmet need of a therapeutic agent to treat SSc-DU by utilizing the pleiotropic effect of riociguat on vascular remodeling and anti-fibrotic, anti-proliferative effect, and anti-inflammatory effects [10,11,12,13,14,15]. We did not find statistically significant improvements in the primary and secondary outcome measures, including patient-reported symptoms of RP and PROs. There are several reasons for this negative trial. First, the trial design excluded PDE5 inhibitors due to relative contraindication of PDE5 inhibitors and riociguat (both target nitric oxide pathway and risk of significant hypotension). The trial allowed background stable CCBs, ACE inhibitors, and anti-platelet therapies. PDE5 inhibitors have become the mainstay for the management of SSc-DU, especially those who do not respond or are intolerant to CCBs [19]. This likely led to the recruitment of a population with milder burden of digital disease—the mean number of active and painful intermediate DUs at baseline was 2.6. Second, the participants in the trial had longer disease duration (mean [SD] 12.0 [10.1] years) compared to the recently completed RISE-SSc trial—a phase 2b randomized controlled trial in participants with early dcSSc (disease duration of mean 9 months) where riociguat was associated with trends in improvement in RP at 14 weeks and DUs at 52 weeks [17]. Although the participants in the placebo group had longer disease duration (15 [8.2] years vs 6.2 [5.8] years), both groups of patients may have had chronic irreversible vasculopathy that was not amenable to oral therapy like riociguat. Third, the duration of riociguat treatment in the RESCUE study, especially with an 8-week titration to maximum tolerated dose, may be suboptimal for healing of DUs. In the open-label extension phase, all baseline and cardinal ulcers in the riociguat-riociguat arm. This observation is a likely indication that a longer duration of riociguat treatment can cause healing of DUs. A similar observation was demonstrated in a RCT of oral treprostinil where the primary outcome of change in net DU burden was not met at week 20 but showed efficacy during the 1-year open-label extension phase when exposed to oral treprostinil [20]. Also, an increase in DU burden was noted in the year after oral treprostinil discontinuation despite adjustment to seasonal variations.
The biomarkers that we chose have been shown to be associated with SSc vasculopathy [21]. At baseline, there were statistically significant elevations in plasma cGMP, sE-selection, and sICAM1 in participants compared to healthy controls, which coincides with the published literature [22,23,24]. After riociguat treatment, none of the biomarkers measured was significantly altered, except for cGMP, which is expected, and supports the target engagement.
Strengths and limitations of the study
We included experienced centers with specialized expertise in the management of SSc-related DUs. The wound care of the DUs was standardized across the participating sites. All DUs were defined, photographed, and confirmed by the corresponding author, suggesting a level of standardization in DU assessment. The study is not without limitations. First, we had a small sample size as this was a pilot study to obtain preliminary estimates of treatment effects in efficacy and safety. We also stopped the trial prematurely due to difficulty in recruitment, as majority of current management of DU in the USA includes PDE5 inhibitors. Second, there was a baseline imbalance in the study population with the placebo group having participants with longer disease duration, but the participants in the riociguat group had more severe self-reported disease in terms of RP and DU.
Conclusions
In conclusion, treatment with riociguat in this trial did not reduce the NUB in patients with SSc. The negative results may reflect lack of power, low NUB at baseline, moderate-to-severe vasculopathy with long-term disease, shorter duration of the trial, and difficulty to recruit patients in the era of widespread use of PDE5 inhibitors. This and other recent trials also highlight the changing epidemiology of SSc-DU due to the availability of somewhat effective pharmacologic therapies such as PDE5 inhibitors, prostacyclin analogs, and better wound care management of these ulcers [20, 25, 26]. Future trials should acknowledge this during the trial design and plan longer trials with background standard of care treatments. There was a trend toward DU healing with longer duration of treatment with riociguat in the open-label extension, but this observation will need to be confirmed in a larger RCT.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- SSc:
-
Systemic sclerosis
- RP:
-
Raynaud’s phenomenon
- DU:
-
Digital ulcers
- PDE5:
-
Phosphodiesterase
- sGC:
-
Soluble guanylate cyclase
- NO:
-
Nitric oxide
- DCC:
-
Data Coordinating Center
- MCPs:
-
Metacarpophalangeal joints
- FRP:
-
Females of reproductive potential
- RCS:
-
Raynaud’s Condition Score
- HAQ-DI:
-
Health Assessment Questionnaire Disability Index
- PROMIS:
-
Patient-Reported Outcomes Measures Information System
- HDISS-DU:
-
Hand Disability in Systemic Sclerosis-DU
- VAS:
-
Visual analog scale
- SHAQ:
-
Scleroderma Health Assessment Questionnaire
- VEGF:
-
Vascular endothelial growth factor
- tPA:
-
Tissue plasminogen activator
- sE-Selectin:
-
Soluble E-selectin
- bFGF:
-
Basic fibroblast growth factor
- VCAM-1:
-
Vascular cell adhesion molecule-1
- sICAM-1:
-
Soluble intracellular adhesion molecule 1
- PINP:
-
N-terminal propeptide of type I collagen
- MMP12:
-
Matrix metalloproteinase 12
- cGMP:
-
Cyclic guanylyl cyclase
- sFLT1:
-
Soluble fms-like tyrosine kinase-1
- MITT:
-
Modified intention to treat
References
Varga J, Trojanowska M, Kuwana M. Pathogenesis of systemic sclerosis: recent insights of molecular and cellular mechanisms and therapeutic opportunities. J Scleroderma Relat Disord. 2017;2(3):137–52.
Seibold JR, Wigley FM, Schiopu E, Denton CP, Silver RM, Steen VD, et al. Digital ulcers in Ssc treated with oral treprostinil: a randomized, double-blind, placebo-controlled study with open-label follow-up. J Scleroderma Relat Disord. 2017;2(1):42–9.
Hughes M, Pauling JD. Exploring the patient experience of digital ulcers in systemic sclerosis. Semin Arthritis Rheum. 2019;48(5):888–94.
Tingey T, Shu J, Smuczek J, Pope J. Meta-analysis of healing and prevention of digital ulcers in systemic sclerosis. Arthritis Care Res (Hoboken). 2013;65(9):1460–71.
Stasch JP, Pacher P, Evgenov OV. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation. 2011;123(20):2263–73.
Grimminger F, Weimann G, Frey R, Voswinckel R, Thamm M, Bölkow D, et al. First acute haemodynamic study of soluble guanylate cyclase stimulator riociguat in pulmonary hypertension. Eur Respir J. 2009;33(4):785–92.
Coghlan JG, Galie N, Barbera JA, Frost AE, Ghofrani HA, Hoeper MM, et al. Initial combination therapy with ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): subgroup analysis from the AMBITION trial. Ann Rheum Dis. 2017;76(7):1219-27.
Ghofrani HA, Hoeper MM, Halank M, Meyer FJ, Staehler G, Behr J, et al. Riociguat for chronic thromboembolic pulmonary hypertension and pulmonary arterial hypertension: a phase II study. Eur Respir J. 2010;36(4):792–9.
Ghofrani HA, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369(4):330–40.
Sharkovska Y, Kalk P, Lawrenz B, Godes M, Hoffmann LS, Wellkisch K, et al. Nitric oxide-independent stimulation of soluble guanylate cyclase reduces organ damage in experimental low-renin and high-renin models. J Hypertens. 2010;28(8):1666–75.
Lang M, Kojonazarov B, Tian X, Kalymbetov A, Weissmann N, Grimminger F, et al. The soluble guanylate cyclase stimulator riociguat ameliorates pulmonary hypertension induced by hypoxia and SU5416 in rats. PLoS One. 2012;7(8):e43433.
Dees C, Beyer C, Distler A, Soare A, Zhang Y, Palumbo-Zerr K, et al. Stimulators of soluble guanylate cyclase (sGC) inhibit experimental skin fibrosis of different aetiologies. Ann Rheum Dis. 2015;74(8):1621–5.
Distler O, Pope J, Denton C, Allanore Y, Matucci-Cerinic M, de Oliveira Pena J, et al. RISE-SSc: riociguat in diffuse cutaneous systemic sclerosis. Respir Med. 2017;122 Suppl 1:S14–S7.
Beyer C, Reich N, Schindler SC, Akhmetshina A, Dees C, Tomcik M, et al. Stimulation of soluble guanylate cyclase reduces experimental dermal fibrosis. Ann Rheum Dis. 2012;71(6):1019–26.
Beyer C, Zenzmaier C, Palumbo-Zerr K, Mancuso R, Distler A, Dees C, et al. Stimulation of the soluble guanylate cyclase (sGC) inhibits fibrosis by blocking non-canonical TGFβ signalling. Ann Rheum Dis. 2015;74(7):1408–16.
Huntgeburth M, Kießling J, Weimann G, Wilberg V, Saleh S, Hunzelmann N, et al. Riociguat for the treatment of Raynaud’s phenomenon: a single-dose, double-blind, randomized, placebo-controlled cross-over pilot study (DIGIT). Clin Drug Investig. 2018;38(11):1061–9.
Khanna D, Allanore Y, Denton CP, Kuwana M, Matucci-Cerinic M, Pope JE, et al. The effects of riociguat on Raynaud’s phenomenon and digital ulcers in patients with diffuse systemic sclerosis: results from the Phase IIb RISE-SSc Study. In: Arthritis & rheumatology. NJ USA: WILEY 111 RIVER ST, HOBOKEN 07030-5774; 2018.
van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Arthritis Rheum. 2013;65(11):2737–47.
Kowal-Bielecka O, Fransen J, Avouac J, Becker M, Kulak A, Allanore Y, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis. 2017;76(8):1327–39.
Shah AA, Schiopu E, Chatterjee S, Csuka ME, Frech T, Goldberg A, et al. The recurrence of digital ulcers in patients with systemic sclerosis after discontinuation of oral treprostinil. J Rheumatol. 2016;43(9):1665–71.
Chora I, Guiducci S, Manetti M, Romano E, Mazzotta C, Bellando-Randone S, et al. Vascular biomarkers and correlation with peripheral vasculopathy in systemic sclerosis. Autoimmun Rev. 2015;14(4):314–22.
Andersen GN, Caidahl K, Kazzam E, Petersson AS, Waldenström A, Mincheva-Nilsson L, et al. Correlation between increased nitric oxide production and markers of endothelial activation in systemic sclerosis: findings with the soluble adhesion molecules E-selectin, intercellular adhesion molecule 1, and vascular cell adhesion molecule 1. Arthritis Rheum. 2000;43(5):1085–93.
Yalçınkaya Y, Adın-Çınar S, Artim-Esen B, Kamalı S, Pehlivan Ö, Öcal L, et al. Capillaroscopic findings and vascular biomarkers in systemic sclerosis: association of low CD40L levels with late scleroderma pattern. Microvasc Res. 2016;108:17–21.
Cossu M, Andracco R, Santaniello A, Marchini M, Severino A, Caronni M, et al. Serum levels of vascular dysfunction markers reflect disease severity and stage in systemic sclerosis patients. Rheumatology (Oxford). 2016;55(6):1112–6.
Khanna D, Denton CP, Merkel PA, Krieg T, Le Brun FO, Marr A, et al. Effect of macitentan on the development of new ischemic digital ulcers in patients with systemic sclerosis: DUAL-1 and DUAL-2 randomized clinical trials. JAMA. 2016;315(18):1975–88.
Hachulla E, Hatron PY, Carpentier P, Agard C, Chatelus E, Jego P, et al. Efficacy of sildenafil on ischaemic digital ulcer healing in systemic sclerosis: the placebo-controlled SEDUCE study. Ann Rheum Dis. 2016;75(6):1009–15.
Acknowledgements
Not applicable
Funding
This is an investigator-initiated trial designed by the sponsor (Dinesh Khanna). The study was funded by a grant of Bayer AG (Leverkusen) and Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. The companies had no influence on data interpretation and reporting of the trial. The data was stored at the University of Michigan Data Coordinating Center. The manuscript was drafted by Vivek Nagaraja, Cathie Spino, and Dinesh Khanna with input from other co-authors. No medical writer was involved in the creation of the manuscript. The manuscript was reviewed by the Bayer before final submission. The corresponding author had full access to all data congregates in the study and made the final decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Contributions
All authors drafted the article, revised it critically for important intellectual content, approved the final version to be published. DK and CS had full access to all the data in the study, take responsibility for the integrity of the data and the accuracy of the data analysis, and were responsible for the study conception and design and analysis and interpretation of the data. VN, RD, RL, TF, JG, VS, and DK were responsible for the acquisition of the data.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Each site’s institutional review board or ethics committee approved the protocol before the study commenced. The study was conducted in accordance with the principles of the Declaration of Helsinki.
Consent for publication
Not applicable
Competing interests
Dr. Robyn Domsic has worked as a consultant for paid consultant for Eicos Sciences Inc. and Boehringer-Ingelheim. Dr. Robert Lafyatis has received grant support from PRISM Biolab, Regeneron, Elpidera, and Kiniksa. He has served as a consultant for PRISM Biolab, Merck, Bristol Myers Squibb, Biocon, UCB, Formation, Sanofi, and Genentech/Roche. Dr. Jessica Gordon has received grant support from Corbus Pharmaceuticals and Cumberland Pharmaceuticals. Dr. Dinesh Khanna has served as a consultant for Actelion, Acceleron, BMS, Blade Therapeutics, Boehringer Ingelham, Bayer, ChemomAB, Cytori, Celgene, Curzion, Corbus Pharmaceuticals, CSL Behring, GSK, Genentech, Mitsubishi Tanabe Pharma Development America, Sanofi-Aventis, and UCB. He has stocks in Eicos Sciences, Inc. and has employment with CiviBio Pharma, Inc. He has received grant support from Bristol Myers Squibb, Pfizer, Bayer, and Horizon. The rest of the authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Complete inclusion/exclusion criteria. (PDF 904 kb)
Additional file 2:
Further details on the statistical analysis. (PDF 681 kb)
Additional file 3:
Comparison of the statsitical change in biomarkers levels from baseline between healthy controls and all patients. (DOCX 23 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nagaraja, V., Spino, C., Bush, E. et al. A multicenter randomized, double-blind, placebo-controlled pilot study to assess the efficacy and safety of riociguat in systemic sclerosis-associated digital ulcers. Arthritis Res Ther 21, 202 (2019). https://doi.org/10.1186/s13075-019-1979-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-019-1979-7