
Abstract
Background
Immune checkpoint blockade (ICB) with antibodies inhibiting cytotoxic T lymphocyte-associated protein-4 (CTLA-4) and programmed cell death protein-1 (PD-1) (or its ligand (PD-L1)) can stimulate immune responses against cancer and have revolutionized the treatment of tumors. The influence of host germline genetics and its interaction with tumor neoantigens remains poorly defined. We sought to determine the interaction between tumor mutational burden (TMB) and the ability of a patient’s major histocompatibility complex class I (MHC-I) to efficiently present mutated driver neoantigens in predicting response ICB.
Methods
Comprehensive genomic profiling was performed on 83 patients with diverse cancers treated with ICB to determine TMB and human leukocyte antigen-I (HLA-I) genotype. The ability of a patient’s MHC-I to efficiently present mutated driver neoantigens (defined by the Patient Harmonic-mean Best Rank (PHBR) score (with lower PHBR indicating more efficient presentation)) was calculated for each patient.
Results
The median progression-free survival (PFS) for PHBR score < 0.5 vs. ≥ 0.5 was 5.1 vs. 4.4 months (P = 0.04). Using a TMB cutoff of 10 mutations/mb, the stable disease > 6 months/partial response/complete response rate, median PFS, and median overall survival (OS) of TMB high/PHBR high vs. TMB high/PHBR low were 43% vs. 78% (P = 0.049), 5.8 vs. 26.8 months (P = 0.03), and 17.2 months vs. not reached (P = 0.23), respectively. These findings were confirmed in an independent validation cohort of 32 patients.
Conclusions
Poor presentation of driver mutation neoantigens by MHC-I may explain why some tumors (even with a high TMB) do not respond to ICB.
Similar content being viewed by others


Background
Immune checkpoint blockade (ICB) with antibodies inhibiting cytotoxic T lymphocyte-associated protein-4 (CTLA-4) and programmed cell death protein-1 (PD-1) (or its ligand (PD-L1)) can stimulate immune responses against cancer and has revolutionized the treatment of both solid [1] and hematologic malignancies [2]. Durable remissions after ICB have been reported in patients with diverse advanced cancers including, but not limited to, melanoma [3], non-small cell lung cancer (NSCLC) [4], renal cell carcinoma [5], and Hodgkin lymphoma [6]. Still, responses to ICB can be variable, toxicity can be serious, resistance is common [7], and hyperprogression can occur [8]. Further, the majority of patients will not benefit from ICB, and there is a need to better select patients for treatment [9].
Multiple factors influence the immune response against tumors including tumor T cell infiltration, tumor mutational burden (TMB), PD-L1 expression, interferon signaling, mismatch repair (MMR) deficiency, tumor aneuploidy, and possibly the intestinal microbiota [10]. Biomarkers that have entered clinical practice include PD-L1 expression measured by immunohistochemistry (IHC) [11], PD-L1 amplification [12], microsatellite instability (MSI) [13, 14], and TMB [15, 16].
Somatic mutations in tumors can be recognized by the immune system [17] resulting in tumor eradication. MMR-deficient/MSI-high tumors have 10 to 100 times as many somatic alterations as MMR-proficient tumors [13], resulting in exquisite sensitivity to ICB therapy [14]. Most cancers harboring MMR alterations are associated with high TMB [18]. In addition, many cancers harbor high TMB (10–20% depending on the definition of high TMB), even without MMR alterations [15, 19]. Higher TMB correlates with better treatment outcomes, including higher response rates and longer progression-free survival (PFS) and overall survival (OS), in diverse cancers treated with immunotherapies [15].
Despite the improved efficacy of ICB in TMB-high tumors, approximately 40–60% of patients with a high TMB will not respond [15, 16]. To date, there is no sufficient way to predict which patients with high TMB will or will not respond to ICB. It has been hypothesized that tumors with high TMB and low PD-L1 expression might not respond as well to ICB; however, studies have demonstrated higher response rates and PFS in patients with high TMB versus low TMB, irrespective of PD-L1 expression [20].
Major histocompatibility complex class I (MHC-I) molecules, encoded by the human leukocyte antigen-I (HLA-I) locus, present intracellular peptides on the surface of both normal and tumor cells for recognition by CD8+ cytotoxic T cells [21]. HLA-I genotype has been linked to a variety of different immune responses including infection [22], autoimmune diseases [23], and the graft versus host/tumor effect seen after allogeneic stem cell transplantation [24]. There is accumulating experimental evidence suggesting that immunosurveillance shapes the mutational landscapes of cancers through the elimination of early tumor cells [25,26,27]. In addition, the predicted number of MHC-I-associated neoantigens has been shown to be low in certain tumors suggesting immune-mediated elimination [28], and the anti-tumor activity of ICB is dependent on MHC-I presentation of specific tumor-derived peptides [29, 30].
Marty et al. developed a residue-centric patient MHC-I presentation score (termed the Patient Harmonic-mean Best Rank (PHBR) score) that describes a person’s ability to present specific cancer mutations to CD8+ T cells, and found that PHBR scores correlated with the likelihood of mutations to emerge in a patient’s tumor [31]. Poor presentation of a mutation across patients was correlated with higher frequency among tumors. These results support that MHC-I genotype-restricted immunoediting shapes the mutational landscape of malignancies.
It has been suggested that the presence of a high-quality neoantigen is required for response to therapy [32] while a high burden of neoantigens has been associated with impaired anti-tumor immune activity [33]; thus, we focused on neoantigen quality over quantity by using patient minimum PHBR score (i.e., best-presented mutation) to predict whether mutations observed in a patient’s tumor are likely to generate effectively presented neoantigens. We assessed the ability of PHBR and TMB to predict response to ICB in diverse solid tumors.
Methods
Patient selection
Three hundred and twenty-eight patients with diverse solid tumors treated with ICB (4/2010–5/2018) at a single institution were reviewed. Patients with melanoma, tumors that were not sequenced by Foundation Medicine (FM), and patients without an identified missense alteration by NGS were excluded. We excluded patients without next-generation sequencing or those with sequencing, but no identified missense alterations, because PHBR cannot be calculated in those cases; we omitted melanoma because melanoma patients have disproportionately high TMBs and high response rates to immunotherapy as compared to the majority of other cancers. All patients were treated with anti-PD-1/L1 monotherapy (or in combination with a second agent). The validation cohort was composed of thirty-two NSCLC patients treated with pembrolizumab (starting from 2012 to 2013) at Memorial Sloan Kettering and the University of California Los Angeles. All validation patients had consented to Institutional Review Board-approved protocols regarding tissue collection and sequencing.
TMB and HLA-I sequencing
Patients had NGS performed on tumor samples to determine genetic alterations, TMB, and HLA-I genotype [34]. Formalin-fixed paraffin-embedded tumor samples were submitted for NGS to FM [clinical laboratory improvement amendments (CLIA)-certified lab]. The FoundationOne assay was used (hybrid-capture-based NGS; 236 or 315 genes; http://www.foundationone.com/). The methods have been previously described [34]. Average sequencing depth of coverage was greater than 250X, with > 100X at > 99% of exons. For TMB, the number of somatic mutations detected on NGS (interrogating 1.2 mb of the genome) is quantified and that value extrapolated to the whole exome using a validated algorithm [35]. Alterations likely or known to be bona fide oncogenic drivers and germline polymorphisms are excluded. TMB was measured in mutations per megabase (mb). Sequence-derived HLA-A/B/C typing was conducted by back-converting BAM files to fastq, then performing HLA realignment and typing using OptiType [36].
PHBR
The Patient Harmonic-mean Best Rank (PHBR) score as previously described [31], is a metric that represents how well the specific HLA-I genotype of an individual can bind and present a specific missense mutation. Each patient was assigned the PHBR score of his or her best-presented missense driver mutation. For patients with two or more missense mutations, only the mutation with the lowest PHBR score was selected. PHBR low (strong presentation) and high (poor presentation) were defined as < 0.5 and ≥ 0.5, respectively.
Mapping Foundation Medicine mutations to peptides
RefSeq transcript IDs from the FM variant spreadsheet were mapped to corresponding Ensembl transcript IDs with coding (CDS) sequences. For evaluation of missense mutations, we replaced the native amino acid residue with the mutated residue and selected all 38 possible peptides of length 8–11 that covered the mutated amino acid residue. For evaluation of in-frame insertion and deletion mutations, bases were inserted or deleted from the CDS sequence according to the “cds effect” column from the FM data. The new CDS sequence was then translated into an amino acid sequence using the Seq.translate function from Biopython (Bio) package [37]. We then selected any resulting novel peptides of length 8–11 for affinity analysis.
Affinity analysis
We calculated the allele-specific binding affinities of the previously described mutated peptides using NetMHCpan4.0 [38]. Conventionally, a NetMHCpan4.0 binding affinity percentile rank less than 2 indicates weak peptide-MHC binding, while a binding affinity percentile rank less than 0.5 indicates strong peptide-MHC binding [39]. Patient Harmonic-mean Best Rank PHBR scores [31] were used to represent a patient’s ability to present the mutations in their tumor. HLA-A, HLA-B, and HLA-C alleles were obtained from FM. We evaluated the binding affinity of each HLA allele for 38 possible peptides of length 8–11 overlapping each mutation using NetMHCpan4.0. For individual alleles, the best rank percentile from NetMHCpan4.0 out of the 38 possible peptides was assigned. Best rank percentiles for all 6 alleles were aggregated into the PHBR score using a harmonic mean. High PHBR scores are indicative of poor affinity of peptides overlapping a mutation with the patient’s MHC-I molecules and vice versa.
Validation
Matched tumor-normal exome sequencing fastq files obtained from [40] (dbGaP study accession phs000980.v1.p1.c1) were preprocessed and mutations called according to the GATK best practice workflow. Only mutations occurring in the 309 genes from the Foundation Medicine gene panel were retained. HLA typing was done in silico using the OptiType software package [41]. Mutated peptides were created using the same method as described above. Similarly, PHBR scores were generated as described previously.
Statistical analysis
We used the Fisher exact test to assess categorical variables. P values < 0.05 were considered significant (values < 0.10 were included in the multivariable regression analyses). Overall benefit rate (OBR) (stable disease for ≥ 6 months and partial or complete response) was determined (RECIST criteria). Median PFS and OS were calculated from the start of checkpoint blockade and data was censored at the last visit for patients still progression free or alive, respectively, for PFS and OS. For the outcome analysis, comparisons were made between TMB low vs. high and PHBR low vs. high. Patients with no TMB values were assigned to the low TMB category for discrete analyses, and a pseudocount of 0.001 was added to TMB for all patients. We performed a Cox proportional hazards regression stratified by high (≥ 10 mutations/mb) or low (< 10 mutations/mb) TMB to quantify the specific effect of PHBR on PFS. These findings were visualized using Kaplan-Meier curves. Statistical analysis was performed on R version 3.5.2 and IBM SPSS Statistics version 24.
Results
Eighty-three patients with 20 different solid malignancies were identified (Table 1, Additional file 1: Table S1, and Additional file 1: Fig. S1, and Additional file 1: Fig. S2). The most common malignancies in the cohort included non-small cell lung cancer (NSCLC) (N = 26), cutaneous squamous cell carcinoma (SCC) (N = 10), and head and neck SCC (N = 9). Sixty-six patients were treated with PD-1/L1 monotherapy and 17 with combination therapy. The OBR (stable disease (SD) ≥ 6 months/partial and complete response (PR/CR)) was 43%. Thirty-two patients had at least one PHBR score of < 0.5 and fifty-one patients had a minimum score ≥ 0.5 (lower scores reflecting better neoantigen presentation) (Additional file 1: Fig. S3). A minimum PHBR score ≥ 0.5 was significantly associated in univariate analysis with progressive disease (P = 0.02), non-cutaneous SCC malignancies (P = 0.04), and a TMB < 50 mutations/mb (P = 0.05).
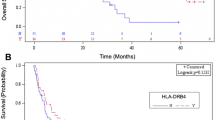
In univariate analysis (Table 2), only higher TMB (≥ 10 mutations/mb) was associated with a better OBR. Caucasian ethnicity, high TMB, and a minimum PHBR score < 0.5 were all significantly associated with longer median PFS while male sex, Caucasian ethnicity, and high TMB were associated with longer median OS. The median PFS for low versus high PHBR scores was 5.1 vs. 4.4 months (P = 0.04) (Fig. 1). The median PFS for high versus low TMB at various thresholds (10, 20, 50) was 6.9 vs. 4.0 months (P = 0.001), 14.1 vs. 4.2 months (P = 0.01), and 26.8 vs. 4.4 months (P = 0.03), respectively.

Kaplan-Meier PFS and OS for patients treated with immunotherapy. P values in Fig. 1 compare all four categories. They differ slightly from P values in Table 3, which compares value to the reference. PFS (a) and OS (b) dichotomized by PHBR < 0.5 and ≥ 0.5 (N = 83). PFS (c) and OS (d) dichotomized by TMB < 10 and ≥ 10 mutations/mb (N = 83). PFS (e) and OS (f) separated by TMB < 10 and ≥ 10 and PHBR < 0.5 and ≥ 0.5 (N = 83). For PFS (e), P = 0.005 for difference between all four curves. Curve for TMB ≥ 10/PHBR < 0.5 versus TMB ≥ 10/PHBR ≥ 0.5 was significantly different (P = 0.025); TMB ≥ 10/PHBR ≥ 0.5 did not differ significantly from TMB < 10/PHBR ≥ 0.5 (P = 0.19) or from TMB < 10/PHBR < 0.5 (P = 0.26); TMB < 10/PHBR ≥ 0.5 did not differ significantly from TMB < 10/PHBR < 0.5 (P = 0.91). For OS (f), P = 0.1 for difference between all four curves. Differences between individual curves were not statistically different
Using a TMB cutoff of 10 mutations/mb, the OBR, median PFS, and median OS of TMB low/PHBR high vs. TMB high/PHBR low were 33% vs. 78% (P = 0.006), 3.5 vs. 26.8 months (P < 0.001), and 10.1 months vs. not reached (P = 0.008), respectively (Fig. 1 and Table 3). Results remain when we exclude patients who had unknown TMB values (Additional file 1: Fig. S4). Patients with high TMB had greater OBR (43% vs. 78%, P = 0.049), greater PFS (5.8 vs. 26.8 months, P = 0.03), and greater median overall survival (17.2 months vs. not reached, P = 0.23) when accompanied by a well-presented mutation (low PHBR) than their counterparts with less well-presented mutations (high PHBR) (Table 3, Additional file 1: Fig. S5).
In a multivariable regression analysis (Table 4) of factors affecting outcome for patients treated with immunotherapy, high TMB (P = 0.01) and treatment with combination therapy (P = 0.006) were significantly associated with a higher OBR. Only high TMB was significantly associated with a prolonged median PFS (P = 0.01) and OS (P = 0.04). However, in stratified Cox regression, which allows for different hazard functions among strata [42] of PHBR in the higher TMB (≥ 10 mutations/mb) patients (N = 39), we found that a low PHBR score is significantly predictive of PFS (HR 0.39 (0.16–0.91), P = 0.03). Multivariable regression analysis in this cohort of 39 patients with high TMB showed that PHBR, but not TMB, was selected as an independent factor predicting both OBR and longer PFS (P = 0.049 and 0.03, respectively) (Additional file 1: Table S2 and Table S3). In contrast, PHBR had no effect on PFS (P = 0.98) in patients with lower TMB (< 10 mutations/mb) (N = 38). Plotting Kaplan-Meier curves of patients based on lower or higher TMB and low or high PHBR found similar results in the general cohort (i.e., PHBR low versus high is associated with significant separation of the curves in patients with TMB ≥ 10 mutations/mb, but not in patients with lower TMBs (Fig. 1)). Finally, overall, Spearman correlation coefficient between TMB and PHBR was 0.31 with a P value of 0.01, consistent with a higher likelihood of carrying a low PHBR mutation when TMB is high (Additional file 1: Fig. S6).
Next, we evaluated the added value of PHBR with respect to TMB from another perspective. We first fit a logistic regression model relating OBR to all potential confounders, using a backward selection process where we removed confounders one at a time and compared models using Akaike Information Criterion (AIC) scores [43]. We kept all confounders for which exclusion did not result in an increased AIC (i.e., the model better explained the data when the confounder was included). The retained confounders included MSI status, ethnicity, and the type of cancer each patient was diagnosed with. Then, we sequentially added TMB and PHBR to the regression model, using AIC once again to compare models (Table S8). We found that with the confounders and TMB in the model, the addition of the PHBR results in a reduction of AIC, indicating added explanatory power of PHBR even when TMB is included. In the final model with all the selected confounders, TMB and PHBR, the PHBR has a negative coefficient with a P value of 0.08. The AUC values associated with the final models with confounders were 0.64 for both TMB and PHBR models alone, and 0.68 for the model with both TMB and PHBR (Additional file 1: Fig. S7).
To investigate the generalizability of our analyses across histologies, we revisited Kaplan-Meier analysis for progression-free survival within tumor types with at least 5 patients (NSCLC, SCC, head and neck, breast) (Additional file 1: Fig. S8) and in all tumors excluding NSCLC and SCC, the two most common histologies (Additional file 1: Fig. S9). In each of these analyses, we observed that low versus high PHBR similarly stratified patients with high TMB. In addition, when we train a logistic regression classifier using the two most frequent histologies (N = 31), NSCLC and SCC, and predict response for the remaining patients (N = 46), we observe that the combination of PHBR and TMB better predicts OBR (Additional file 1: Fig. S10). These results suggest that the information provided by TMB and PHBR generalizes beyond high mutation burden tumors such as SCC and NSCLC.
In an external validation cohort of 32 patients with NSCLC treated with pembrolizumab (Additional file 1: Table S4, Table S5 and Fig. S3), the results were similar to those in our UCSD cohort: the OBR and median PFS of PHBR < 0.5 vs. ≥ 0.5 was 76% vs. 30% (P = 0.02) and 14.5 vs. 2.1 months (P < 0.001), respectively (Fig. 2, Additional file 1: Table S6). Using a TMB cutoff of 10 mutations/mb, the median PFS of TMB high/PHBR high vs. TMB high/PHBR low was 8.1 months, versus not reached, respectively (P = 0.02) (Fig. 2, Additional file 1: Table S7). OS data was not available for analysis.

PFS for patients treated with immunotherapy in the validation dataset (N = 32). P values in the figure compare all four categories. a PFS dichotomized by PHBR < 0.5 and ≥ 0.5. b PFS dichotomized by TMB < 10 and ≥ 10 mutations/mb. c PFS separated by TMB < 10 and ≥ 10 and PHBR < 0.5 and ≥ 0.5
Finally, we compared our findings in an aggregated high-TMB melanoma cohort [44,45,46,47] and a low TMB kidney cancer cohort [48]. While minimum PHBR score did not significantly stratify melanoma patient overall or progression-free survival across all patients (Fig. 3a, b), we did find, when also considering sex and age, that lower PHBR scores (i.e., better presented mutations) were significantly associated with better overall and progression-free survival outcomes in high-TMB patients (Table 5), consistent with our reported findings. As expected in the low TMB kidney tumors, there was no correlation between mutation burden and increased progression-free or overall survival (Fig. 4a, b). Interestingly, while we did not see significant survival stratification with min-PHBR (Fig. 4c, d), we did find that responders tended to have lower PHBR scores (i.e., better presented mutations) than non-responders, although the trends did not reach statistical significance (Fig. 5).

Kaplan-Meier curves showing the effects of a TMB on overall survival, b TMB on progression-free survival, and c minimum PHBR score on overall survival and d minimum PHBR score on progression-free survival in the combined melanoma cohort

Kaplan-Meier curves showing the effects of a TMB on overall survival, b TMB on progression-free survival, and c minimum PHBR score on overall survival and d progression-free survival in the Miao kidney cohort

Boxplots showing the distribution of a TMB and b minimum PHBR score for responders and non-responders in the Miao cohort. P values were calculated by the Mann-Whitney U test
Discussion
In a cohort of 83 patients with diverse solid tumors, we demonstrate that both TMB and efficient neoantigen presentation (defined by at least one PHBR score < 0.5) predict better response (as defined by SD ≥ 6 months/PR/CR rate) and longer PFS and OS after treatment with ICB. This finding was confirmed in an independent cohort of 32 patients with NSCLC treated with PD-1 blockade. Further, by incorporating the PHBR score, we were able to identify a group of higher TMB tumors (≥ 10 mutations/mb) that are less likely to benefit from ICB. Specifically, patients with tumors that poorly present driver neoantigens are less likely to respond to ICB, even in tumors with a higher mutational load. Numerous studies show that a significant proportion of patients with a higher TMB do not respond to ICB and there is a need to better identify this group of patients [15, 16, 19].
Chowell et al. demonstrated that HLA-I homozygosity and somatic loss of heterozygosity (LOH) are predictive of poor outcomes in two independent cohorts treated with ICB [49]. In addition, McGranahan et al. observed that 40% of early-stage NSCLC tumors had HLA loss of heterozygosity [32]. It was hypothesized that patients homozygous in at least one HLA-I locus would be predicted to present a smaller and less diverse tumor-derived neoantigen repertoire to CD8+ cytotoxic T cells and that the diversity of HLA molecules in a given patient influences the selection and clonal expansion of T cells following ICB [50].
Our report differs from the Chowell et al. in several ways. We assessed patient-specific MHC-I ability to bind to tumor neoantigens (PHBR score), not HLA-I diversity. Furthermore, by evaluating the interaction between TMB and the PHBR score, we demonstrated that tumors that present neoantigens efficiently respond to ICB, at least in the case of higher TMB (≥ 10 mutations/mb). However, in patients with lower TMB, the presentation of neoantigens as reflected by PHBR had no association with outcome. We hypothesize that, when there are multiple neoantigens produced by the mutanome (i.e., in patients with higher TMB), there is the opportunity for MHC-I to present them (or at least one of them) in such a way that is critical to the response. However, when there are few neoantigens, the opportunity to present them may be diminished to such an extent that the PHBR is not impactful. Additional studies will be required to better understand the neoantigen landscape as it relates to host anti-tumor immunity, in addition to the optimal method to combine information across multiple neoantigen for predicting response to therapy.
In our study, all data gathered to identify possible biomarkers to ICB was obtained via one NGS test at one time point. Prediction scores and gene signatures that take into count numerous variables including T cell infiltration into tumors, mutational load, and PD-L1 level have also been developed [51, 52]. Here we show that, with further validation, the PHBR score and TMB obtained via NGS, both of which are easy to assay, provide the ability to deliver data in real time for clinicians to make treatment decisions.
Our study has several limitations. It was a retrospective study that included a non-uniform group of patients with different malignancies treated with different checkpoint inhibitors. However, similar results were obtained in our validation cohort of NSCLC all treated with the same therapy. Our study excluded melanoma and included only small subsets of patients with individual tumor types; while our specific analyses for tumor types with ≥ 5 patients and leave-one-out analyses (Additional file 1: Fig. S8 and Fig. S9) suggest generalizability, much larger sample sizes will be required to determine whether these findings generalize to specific histologies. Our study did not assess T cell receptor (TCR) specificity and diversity. TCR specificity for MHC-I/peptide complex is essential for CD8+ T cell cellular-mediated cytotoxicity. A strong correlation between TCR CDR3 diversity and TMB has been reported [50]. Finally, we only assessed the PHBR score for MHC-I and not MHC-II. MHC-II presentation of neoantigens is possibly an important determinant of an immune response against a tumor. Frequent cancer driver mutations are poorly presented by MHC-II, and MHC-II shows less inter-patient variability but stronger selective effects than MHC-I [53].
Conclusions
In summary, the ability of patient-specific MHC-I complexes to bind and present neoantigens represented by the PHBR score can predict who is most likely to respond to ICB within the subgroup of patients with higher TMB. These results need to be extensively validated prior to incorporation into routine clinical use. Future studies are needed to clarify the role of PHBR score in predicting response to ICB in specific malignancies. Patients with high PHBR scores may benefit from immunotherapies that circumvent antigen presentation by MHC-I (e.g., chimeric antigen receptor T cells). Finally, much effort will be needed to decipher how to best incorporate MHC-I-related PHBR, reflecting neoantigen presentation by HLA-I, in the context of PD-L1 expression, TCR repertoire, and HLA-II genotype.
Availability of data and materials
Clinical data used in this study is included in the online supplement. Whole exome sequencing data used in the validation analyses was downloaded from dbGaP (accession numbers: phs000980.v1.p1.c1 [40], phs001041.v1.p1 [45], phs000452.v2.p1 [46]) and the SRA (project numbers: SRP095809 [44], SRP090294 [47]).
References
Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–86.
Goodman A, Patel SP, Kurzrock R. PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat Rev Clin Oncol. 2017;14:203–20.
Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob J-J, Rutkowski P, Cowey CL, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19:1480–92.
Gettinger S, Horn L, Jackman D, Spigel D, Antonia S, Hellmann M, et al. Five-year follow-up of nivolumab in previously treated advanced non–small-cell lung cancer: results from the CA209-003 study. JCO. 2018;36:1675–84.
Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378:1277–90.
Armand P, Engert A, Younes A, Fanale M, Santoro A, Zinzani PL, et al. Nivolumab for relapsed/refractory classic Hodgkin lymphoma after failure of autologous hematopoietic cell transplantation: extended follow-up of the multicohort single-arm phase II CheckMate 205 trial. J Clin Oncol. 2018;36:1428–39.
Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819–29.
Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin Cancer Res. 2017;23(15):4242–50. https://doi.org/10.1158/1078-0432.CCR-16-3133.
Subbiah V, Kurzrock R. The marriage between genomics and immunotherapy: mismatch meets its match. Oncologist. 2019;24:1–3.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–23.
Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14:847–56.
Goodman AM, Piccioni D, Kato S, Boichard A, Wang H-Y, Frampton G, et al. Prevalence of PDL1 amplification and preliminary response to immune checkpoint blockade in solid tumors. JAMA Oncol. 2018 [cited 2018 Jun 18]; Available from: https://jamanetwork.com/journals/jamaoncology/fullarticle/2684636.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20.
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13.
Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16:2598–608.
Samstein RM, Lee C-H, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–6.
Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, et al. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008;68:889–92.
Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018;7:746–56.
Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30:44–56.
Ready N, HellmannMD, Awad MM, Otterson GA, Gutierrez M, Gainor JF, Borghaei H, Jolivet J, Horn L, Mates M, Brahmer J, Rabinowitz I, Reddy PS, Chesney J, Orcutt J, Spigel DR, Reck M, O'Byrne KJ, Paz-Ares L, Hu W, Zerba K, Li X, Lestini B, Geese WJ, SzustakowskiJD, Green G, Chang H, Ramalingam SS. First-Line Nivolumab Plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer (CheckMate 568): Outcomes by Programmed Death Ligand 1 and Tumor Mutational Burden as Biomarkers. J Clin Oncol. 2019;37(12):992–1000. https://doi.org/10.1200/JCO.18.01042. Epub 2019 Feb 20.
Chowell D, Krishna S, Becker PD, Cocita C, Shu J, Tan X, et al. TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. Proc Natl Acad Sci U S A. 2015;112:E1754–62.
International HIV Controllers Study, Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PIW, et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551–1557.
Gough SCL, Simmonds MJ. The HLA region and autoimmune disease: associations and mechanisms of action. Curr Genomics. 2007;8:453–65.
Kolb H-J. Graft-versus-leukemia effects of transplantation and donor lymphocytes. Blood. 2008;112:4371–83.
DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482:405–9.
Garcia-Lora A, Algarra I, Garrido F. MHC class I antigens, immune surveillance, and tumor immune escape. J Cell Physiol. 2003;195:346–55.
Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400–4.
Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61.
Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–81.
Tran E, Ahmadzadeh M, Lu Y-C, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350:1387–90.
Marty R, Kaabinejadian S, Rossell D, Slifker MJ, van de Haar J, Engin HB, et al. MHC-I genotype restricts the oncogenic mutational landscape. Cell. 2017;171:1272–83 e15.
McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171:1259–71 e11.
Castro A, Ozturk K, Pyke RM, Xian S, Zanetti M, Carter H. Elevated neoantigen levels in tumors with somatic mutations in the HLA-A, HLA-B, HLA-C and B2M genes. BMC Med Genet. 2019;12:107.
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–31.
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Medicine. 2017;9:34.
Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, Kohlbacher O. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics. 2014;30:3310–6.
Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–3.
Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol. 2017;199:3360–8.
Nielsen M, Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016;8:33.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8.
Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33:1152–8.
Therneau T, Grambsch P. Modeling survival data: extending the Cox model. Springer Science & Business MEdia; 2013.
Sakamoto Y, Ishiguro M, Kitagawa G. Akaike information criterion statistics. Dirdecht: D Reidel; 1986.
Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171:934–49 e16.
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–99.
Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–11.
Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44.
Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science. 2018;359:801–6.
Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359:582–7.
Li B, Li T, Pignon J-C, Wang B, Wang J, Shukla SA, et al. Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat Genet. 2016;48:725–32.
Auslander N, Zhang G, Lee JS, Frederick DT, Miao B, Moll T, et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat Med. 2018;24:1545–9.
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24:1550–8.
Marty Pyke R, Thompson WK, Salem RM, Font-Burgada J, Zanetti M, Carter H. Evolutionary pressure against MHC class II binding cancer mutations. Cell. 2018;175:416–28 e13.
Acknowledgements
• phs000980.v1.p1.c1 [40]
◦ We thank the members of the Thoracic Oncology Service and the Chan and Wolchok labs at MSKCC for the helpful discussions. We thank the Immune Monitoring Core at MSKCC, including L. Caro, R. Ramsawak, and Z. Mu, for the exceptional support with processing and banking peripheral blood lymphocytes. We thank P. Worrell and E. Brzostowski for the help in identifying tumor specimens for analysis. We thank A. Viale for the superb technical assistance. We thank D. Philips, M. van Buuren, and M. Toebes for the help in performing the combinatorial coding screens. The data presented in this paper are tabulated in the main paper and in the supplementary materials. This work was supported by the Geoffrey Beene Cancer Research Center (MDH, NAR, TAC, JDW, AS), the Society for Memorial Sloan Kettering Cancer Center (MDH), Lung Cancer Research Foundation (WL), Frederick Adler Chair Fund (TAC), The One Ball Matt Memorial Golf Tournament (EBG), Queen Wilhelmina Cancer Research Award (TNS), The STARR Foundation (TAC, JDW), the Ludwig Trust (JDW), and a Stand Up To Cancer-Cancer Research Institute Cancer Immunology Translational Cancer Research Grant (JDW, TNS, TAC). Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research.
• phs001041.v1.p1 [45]
◦ We thank Martin Miller at the Memorial Sloan Kettering Cancer Center (MSKCC) for his assistance with the NetMHC server, Agnes Viale and Kety Huberman at the MSKCC Genomics Core, Annamalai Selvakumar and Alice Yeh at the MSKCC HLA typing laboratory for their technical assistance, and John Khoury for the assistance in chart review.
Funding
Funded in part by the National Cancer Institute grant P30 CA023100 and the Joan and Irwin Jacobs Fund philanthropic fund. This work was also supported by the NIH grant DP5-OD017937, a CIFAR fellowship, and an Emerging Leader Award from The Mark Foundation for Cancer Research (#18-022-ELA) to H.C., the NIH National Library of Medicine training grant (T15LM011271) to A.C., and the NIH grant TL1TR001443 to P.R.
This study was in part based on the dbGaP dataset phs000980.v1.p1 which was generously made available by investigators at MSKCC with support from the Geoffrey Beene Cancer Research Center, the Society for Memorial Sloan Kettering Cancer Center, Lung Cancer Research Foundation, Frederick Adler Chair Fund, The One Ball Matt Memorial Golf Tournament, Queen Wilhelmina Cancer Research Award, The STARR Foundation, the Ludwig Trust, and a Stand Up To Cancer-Cancer Research Institute Cancer Immunology Translational Cancer Research Grant.
Author information
Authors and Affiliations
Contributions
AMG, HK, and RK designed the study. AG, HK, AC, EDB, and RK wrote the manuscript. AMG, RO, SK, and PR curated and analyzed the clinical data. XZ, RO, AC, and HC performed the statistical analysis. AC, RMK, and HC calculated all the PHRB scores. GF and ES curated and analyzed the data from Foundation Medicine. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Patients analyzed were part of a UCSD Institutional Review Board-approved trial called PREDICT (NCT02478931). The validation cohort [40] was composed of thirty-two NSCLC patients treated with pembrolizumab (starting from 2012 to 2013) at Memorial Sloan Kettering and the University of California Los Angeles according to protocol NCT01295827. All validation patients had consented to Institutional Review Board-approved protocols regarding tissue collection and sequencing. Both protocols permit collection of genomic and related data and its analysis and reporting at any point during the study period. Written consents were obtained, and this study was performed in accordance with UCSD Institutional Review Board guidelines for data analysis and for any investigational treatments. This research conformed to the principles of the Helsinki Declaration.
Consent for publication
Written consent was obtained per protocol for all patients included in this study. Our protocol allows publication of de-identified data.
Competing interests
Dr. Goodman receives speaking fees from Seattle Genetics and consulting fees from Jazz Pharmaceuticals, Daiichi Sankyo, EUSA Pharma, and Kyowa Kirin. Dr. Frampton and Dr. Sokol are employees and equity holders of Foundation Medicine. Dr. Kurzrock receives research funding from Genentech, Merck, Serono, Pfizer, Sequenom, Foundation Medicine, Konica Minolta, Grifols, and Guardant, as well as consultant fees from X Biotech, Loxo, Neomed, and Actuate Therapeutics, and speaker fees from Roche, and has an ownership interest in IDbyDNA and CureMatch Inc. Paul Riviere receives funding from Peptide Logic LLC. Dr. Kato serves as a consultant for Foundation Medicine. Dr. Pyke is an employee of Personalis, Inc. The remaining authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
List of patients who underwent immunotherapy at UCSD (N = 83). Table S2. Univariate analysis of factors affecting outcome for patients with TMB > 10 mutations/mb treated with immune checkpoint blockade (N = 39 with TMB ≥ 10 mutations/mb). Table S3. Multivariate analysis of factors affecting outcome for patients treated with immunotherapy (N = 39 with TMB ≥10 mutations/mb). Table S4. Validation cohort of 32 patients with NSCLC treated with pembrolizumab. Table S5. Validation cohort patient demographics by PHBR score (< 0.5 vs. ≥0.5) for 32 patients with NSCLC treated with pembrolizumab. Table S6. Univariate analysis of factors affecting outcome for validation patients treated with immune checkpoint blockade (N = 32). Table S7. Overall response rate and PFS, segregated by TMB low/high and PHBR low/high among validation patients (N = 32). Table S8. Covariates retained after the backwards selection process. The coefficients and respective p-values for the covariates including TMB and PHBR in the final model are shown. Figure S1. Overview of tumor type distribution for the discovery cohort. Figure S2. CONSORT Diagram. Figure S3. Overview of minimum PHBR score distribution and TMB distribution for the discovery (A-B) and validation (C-D) cohorts. Figure S4. Kaplan and Meier PFS and OS for patients treated with immunotherapy, excluding patients with TMB = 0. Figure S5. Additional PFS and OS for patients treated with immunotherapy (N = 77 with TMB available. Figure S6. Correlation between PHBR score and TMB (N = 77 with TMB available. Figure S7. Area under the receiver operating characteristic curve (AUROC) for predicting OBR in the discovery cohort using the covariates obtained from the backward selection process, with the addition of PHBR (A), TMB (B) and the combination of PHBR and TMB (C). Figure S8. Kaplan Meier PFS dichotomized by both PHBR < 0.5 and ≥ 0.5 and TMB < 10 and ≥ 10 mutations/mb for histologies with ≥5 patients; NSCLC (A), SCC (B), Head and Neck (C), and Breast (D). Figure S9. Kaplan Meier PFS dichotomized by both PHBR < 0.5 and ≥ 0.5 and TMB < 10 and ≥ 10 mutations/mb excluding NSCLC (A), SCC (B), Head and Neck (C), Breast (D) and both NSCLC and SCC, the most common histologies in our cohort (E). Fig. S10: Area under the receiver operating characteristic curve (AUROC) for predicting OBR from PHBR and TMB in the discovery cohort training on NSCLC and SCC patients (A) and testing on patients in the remaining tumor types (B).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Goodman, A.M., Castro, A., Pyke, R.M. et al. MHC-I genotype and tumor mutational burden predict response to immunotherapy. Genome Med 12, 45 (2020). https://doi.org/10.1186/s13073-020-00743-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-020-00743-4