
Abstract
Clinical exome sequencing is frequently used to identify gene-disrupting variants in individuals with neurodevelopmental disorders. While splice-disrupting variants are known to contribute to these disorders, clinical interpretation of cryptic splice variants outside of the canonical splice site has been challenging. Here, we discuss papers that improve such detection.
Similar content being viewed by others
Splicing disruption in human disorders
Gene-disrupting genetic variants frequently lead to neurodevelopmental disorders, including developmental delay and autism spectrum disorder (ASD), when they occur in one of the several hundred genes associated with these disorders [1, 2]. Many of these variants are de novo, observed in the affected child, but not in either parent, and capable of mediating substantial risk for neurodevelopmental disorders. Such variants alter the quantity or quality of the encoded proteins, through deletions, premature stop codons, or missense variants. In this commentary, we consider the impact of an additional class of gene-disrupting variants that act by altering gene splicing. Three papers outline improvements in detecting splice-disrupting variants [3,4,5], and applying these methods predicts cryptic splicing variants in genes associated with neurodevelopmental disorders in about 0.5% of cases and no controls [1, 2].
Splicing motifs and mechanisms
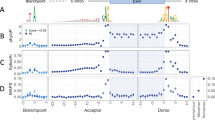
Splicing is a key process in eukaryotic cells. After transcription, a nascent pre-mRNA must be converted into a mature mRNA that can serve as a template for protein translation. This involves the removal of introns from the pre-mRNA, usually by the major spliceosome, through splicing (Fig. 1a). Critical to this process are the two-nucleotide “essential” or “canonical” splice sites (CSS) at either side of exons: an “AG” motif upstream of the acceptor site (A, also called the 3′ splice site), at positions A-1 and A-2, and a “GT” motif downstream of the donor site (D, also called the 5′ splice site), at positions D+1 and D+2 (Fig. 1b).

Overview of the splicing region. a The spliceosome attaches to pre-mRNA as it is transcribed from DNA, removing introns and leaving an exon junction complex upstream. The mature mRNA can migrate out of the nucleus for translation. b Motifs of the polypyrimidine tract, acceptor, and donor, calculated from all protein-coding exons. c Odds ratios of observed and expected variant frequencies around the splice site based on ExAC exome sequencing data in Zhang et al. [4]. Lord et al. [3] use the same ExAC data to calculate the mutability-adjusted proportion of singletons (MAPS) across splicing regions, which is higher at nucleotides intolerant of variation. Jaganathan et al. [5] developed SpliceAI, a neural network for predicting the impact of variants on splicing across the genome; the number of potential variants with a Δ score ≥ 0.1 is shown across splicing regions. Abbreviations: TSS, transcription start site; UTR, untranslated region; A, acceptor; D, donor; Pol II, polymerase II; Ter, termination codon; Poly(A), polyadenylation
Along with the CSS, other DNA features are known to determine splicing behavior, including several motifs representing binding targets of the small nuclear ribonucleoproteins (snRNPs) that make up the major spliceosome. Motif analysis across exons (Fig. 1b) has identified broader “CAG” and “AGGTAAGT” motifs at the acceptor and donor, respectively, as well as the polypyrimidine tract, characterized by enrichment of thymine and cytosine upstream of the acceptor (A-5 to A-40). Upstream of the polypyrimidine tract is the branch point (A-10 to A-50, median A-25) with a “TNA” motif (Fig. 1b). In the major spliceosome, U1 snRNPs bind to the donor site, U2 snRNPs bind to the branch point, and the U2AF protein binds to the polypyrimidine tract and acceptor site [6].
Variation at canonical splice sites
Noncoding genetic variants that disrupt the CSS of critical genes are a known cause of human genetic diseases, including neurodevelopmental disorders [1, 2]. Improper splicing can lead to exon skipping or novel splice sites, both of which can alter the reading frame of protein-coding genes. Alternatively, intron retention incorporates noncoding DNA, which often contains stop codons, into the mature RNA. Consequently, identifying CSS variants in genes with known disease associations is a routine practice in clinical exome sequencing, in which they are treated as protein-truncating variants (PTVs) along with premature stop codons or frameshift variants [7].
Out of 1863 de novo variants identified in individuals with neurodevelopmental disorders [2], the 296 CSS variants account for 16% of PTVs, 637 premature stop codons account for 34%, and 930 frameshift insertions or deletions account for 50%. To quantify the contribution of these de novo variants to disorders, we consider the frequency of variants in 3230 protein-coding genes that are predicted to be “PTV-intolerant,” based on fewer than expected PTVs in whole-exome sequencing data from over 60,000 individuals (expressed statistically as a probability loss-of-function intolerant (pLI) score ≥ 0.9) [8]. Comparing differences in the rate of de novo PTVs in these PTV-intolerant genes between cases and controls [1, 2], we estimate that de novo PTVs contribute to 5% of ASD cases and 16% of developmental delay cases. Since 16% of PTVs are in the CSS, this equates to 0.8% of ASD cases and 2.6% of developmental delay cases due to splicing disruption at the CSS.
Variation at cryptic splice sites
Exonic or intronic splice-disrupting variants outside of the CSS are commonly referred to as cryptic splice variants, due to the challenge of identifying them. The below articles focus on improving clinical interpretation of these cryptic splice sites in neurodevelopmental disorders by leveraging exome sequencing data from population samples [3, 4] or deep learning methods [5]. Zhang et al. [4] use exome sequencing data from over 60,000 population samples from the Exome Aggregation Consortium (ExAC) [8] to assess the observed versus expected number of variants in the 10 nucleotides flanking the acceptor and donor sites in PTV-intolerant genes. They highlight six non-CSS nucleotides that are intolerant of variation (Fig. 1c) and validate splicing dysregulation in four of these (D-1, D+4, D+5, D+6) using paired whole-genome and RNA sequencing from GTEx [9]. Such cryptic splice de novo variants were observed in 0.2% of ASD cases and 0.2% of developmental delay cases. Lord et al. also use ExAC data [3] to highlight two nucleotides that are intolerant of variation (D-1, D+5, Fig. 1c). The D+5 site is also enriched for de novo variants in cases of developmental delay in genes associated with this disorder, as was the polypyrimidine tract when all nucleotides (A-5 to A-25) were considered together. By integrating phenotype data, they identified 18 likely diagnostic de novo variants in 7833 cases (0.2%). Functional assessment of splicing using a minigene assay validated six of the seven likely diagnostic variants that were tested (86%).
Jaganathan et al., which includes the authors of this commentary [5], describe the SpliceAI algorithm, a neural network that predicts the impact of cryptic splice variants based on a pre-mRNA sequence. The network, trained on 10,000 nucleotides of human genomic sequence around 260,000 known splice sites from GENCODE, is used to calculate the SpliceAI Δ score by considering the difference in predicted splicing between reference and variant sequence. Scores range from 0 to 1 with high scores more likely to alter splicing (Fig. 1c). Assessing performance in the paired whole-genome and RNA sequencing data from GTEx [9] identifies splicing disruption proportional to the Δ score (i.e., 20% at 0.2; 80% at 0.8) with higher sensitivity and specificity than prior algorithms [5]. High depth RNA-seq of ASD patient-derived lymphoblastoid cell lines validated 21 of 28 (75%) de novo variants predicted to alter splicing (Δ score, 0.10–0.99; median 0.58), including variants in the ASD-associated genes TCF4 and KDM6B [2]. Of note, analysis of GTEx also revealed widespread tissue-specific splicing, which may lead such validation to underestimate the true accuracy. An excess of de novo variants predicted to alter splicing (Δ score ≥ 0.1) was observed in both developmental delay and ASD, compared to controls. Considering only genes previously associated with neurodevelopmental disorders, de novo variants at cryptic splice sites were observed in 23 out of 3953 ASD cases (0.6%), 21 out of 4293 developmental delay cases (0.5%), and none of the 2073 controls [1, 2].
Overall, SpliceAI predicts about 7-fold more cryptic splice site variants than the other two approaches because it is not limited to specific nucleotides (e.g. D+5), includes splice sites further from the exons, and evaluates each splice site individually. Considering variants assessed consistently between these three methods, SpliceAI predicts all four “likely diagnostic” variants in Lord et al. and 10 of the 18 variants (56%) highlighted by Zhang et al.
With these improvements in detection [3,4,5], we propose that de novo variants at cryptic splice sites identified in exome or genome sequencing of individuals with neurodevelopmental disorders should undergo clinical evaluation in a manner similar to deleterious missense variants. Such evaluation would incorporate evidence from gene association studies, pLI scores, and consistency of phenotype [7].
Prevalence of splicing disruption in neurodevelopmental disorders and therapeutic potential
Using the SpliceAI estimates, splicing disruption by de novo variants in PTV-intolerant genes underlies at least 1.4% of ASD cases (0.8% CSS and 0.6% cryptic, see estimates above) and 3.1% of developmental delay cases (2.6% CSS and 0.5% cryptic, see estimates above). These estimates are equivalent to about 20,000 ASD cases 18 years-of-age or below in the USA and 21,000 equivalent developmental delay cases. Inclusion of more genes (PTV-tolerant, noncoding), whole-genome sequencing to identify deep intronic variants missed by exome sequencing, and consideration of homozygous and heterozygous inherited variation will only increase these estimates.
While splicing variants contribute to thousands of cases of neurodevelopmental disorders, they may offer opportunities for novel therapeutic targets. The success of the FDA-approved antisense oligonucleotide (ASO) Nusinersen to modify splicing behavior, resulting in life-saving clinical improvement in patients with spinal muscular atrophy [10], sets a precedent for treating central nervous system disorders via splicing mechanisms. Such a therapy would need to be developed specifically for each splicing variant in most neurodevelopmental disorders [11]. Key research milestones will include assessing the fraction of splicing variation that can be rescued by ASOs, efficient methods to design and test ASOs, and assessment of the extent of rescue in vivo. These approaches may provide the first insights into whether gene therapy can modify the symptoms of ASD and developmental delay, potentially providing a route to treatment for thousands of individuals with splicing variants and de-risking more complicated approaches to gene therapy that could be applicable in larger populations.
Availability of data and materials
All data are provided by the manuscripts cited in this commentary. SpliceAI Δ scores for SNVs are available at https://basespace.illumina.com/s/5u6ThOblecrh, and the SpliceAI code is available at https://github.com/Illumina/SpliceAI.
Abbreviations
- A:
-
Acceptor
- AI:
-
Artificial intelligence
- ASD:
-
Autism spectrum disorder
- ASO:
-
Antisense oligonucleotide
- CSS:
-
Canonical splice site
- D:
-
Donor
- DNA:
-
Deoxyribonucleic acid
- ExAC:
-
Exome Aggregation Consortium
- FDA:
-
Food and Drug Administration
- GTEx:
-
Genotype-Tissue Expression Project
- KDM6B:
-
Lysine demethylase 6B
- RNA:
-
Ribonucleic acid
- pLI score:
-
Probability loss-of-function intolerant score
- Pol II:
-
Polymerase II
- Poly(A):
-
Polyadenylation
- PTV:
-
Protein-truncating variant
- snRNP:
-
Small nuclear ribonucleoproteins
- TCF4:
-
Transcription factor 4
- Ter:
-
Termination codon
- TSS:
-
Transcription start site
- UTR:
-
Untranslated region
References
McRae JF, Clayton S, Fitzgerald TW, Kaplanis J, Prigmore E, Rajan D, et al. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–8 Available from: http://www.nature.com/doifinder/10.1038/nature21062.
Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, Peng M, Collins R, Grove J, Klei L, Stevens C,Reichert J, Mulhern MS, Artomov M, Gerges S, Sheppard B, Xu X, Bhaduri A, Norman U, Brand H, Schwartz G, Nguyen R, Guerrero EE, Dias C; Autism Sequencing Consortium; iPSYCH-Broad Consortium, Betancur C, Cook EH, Gallagher L, Gill M, Sutcliffe JS, Thurm A, Zwick ME, Børglum AD, StateMW, Cicek AE, Talkowski ME, Cutler DJ, Devlin B, Sanders SJ, Roeder K, Daly MJ, Buxbaum JD. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell. 2020;180(3):568–84.e23. https://doi.org/10.1016/j.cell.2019.12.036. Epub 2020 Jan 23. PubMed PMID: 31981491.
Lord J, Gallone G, Short PJ, McRae JF, Ironfield H, Wynn EH, Gerety SS, He L, Kerr B, Johnson DS, McCann E, Kinning E, Flinter F, Temple IK, Clayton-Smith J, McEntagart M, Lynch SA, Joss S, Douzgou S, Dabir T, Clowes V, McConnell VPM, Lam W, Wright CF, FitzPatrick DR, Firth HV, Barrett JC, Hurles ME; Deciphering Developmental Disorders study. Pathogenicity and selective constraint on variation near splice sites. Genome Res. 2019;29(2):159-70. https://doi.org/10.1101/gr.238444.118. Epub 2018 Dec 26. PubMed PMID: 30587507; PubMed Central PMCID: PMC6360807.
Zhang S, Samocha KE, Rivas MA, Karczewski KJ, Daly E, Schmandt B, et al. Base-specific mutational intolerance near splice sites clarifies the role of nonessential splice nucleotides. Genome Res. 2018;28:968–74.
Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;0:1–14 Elsevier Inc. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0092867418316295.
Fica SM, Nagai K. Cryo-electron microscopy snapshots of the spliceosome: structural insights into a dynamic ribonucleoprotein machine. Nat Struct Mol Biol. 2017;24:791–9.
Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312:1880–7.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91 Nature Publishing Group. Available from: http://www.nature.com/doifinder/10.1038/nature19057.
Aguet F, Brown AA, Castel SE, Davis JR, He Y, Jo B, et al. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–13.
Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377:1723–32 Available from: https://www.ncbi.nlm.nih.gov/pubmed/29091570.
Kim J, Hu C, Moufawad El Achkar C, Black LE, Douville J, Larson A, et al. Patient-customized oligonucleotide therapy for a rare genetic disease. N Engl J Med. 2019;381:1644–52 United States.
Acknowledgements
We are grateful to the groups that provided the exome and genome variant data and RNA-seq data to enable the articles described in this commentary. We thank Claudia Dastmalchi, Lindsay Liang, Kishore Jaganathan, Jeremy McRae, and Sofia Kyriazopoulou Panagiotopoulou for their help with this manuscript.
Funding
S.J.S. was supported by grants from the Simons Foundation (SFARI #647371 and #574598).
Author information
Authors and Affiliations
Contributions
The initial draft of this commentary was written by SJS; all authors reviewed and edited the manuscript. The authors read and approved the final manuscript.
Authors’ information
Not applicable.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
K.K.F. was employed by Illumina at the time of writing this commentary. The following patents related to SpliceAI have been filed: Deep Learning-Based Splice Site Classification, Deep Learning-Based Aberrant Splicing Detection, and Aberrant Splicing Detection Using Convolutional Neural Networks (CNNS). The remaining authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sanders, S.J., Schwartz, G.B. & Farh, K.KH. Clinical impact of splicing in neurodevelopmental disorders. Genome Med 12, 36 (2020). https://doi.org/10.1186/s13073-020-00737-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-020-00737-2