
Abstract
Background
The schizothoracine fishes, an excellent model for several studies, is a dominant fish group of the Qinghai-Tibet Plateau (QTP). However, species populations have rapidly declined due to various factors, and infection with Echinorhynchus gymnocyprii is cited as a possible factor. In the present study, the molecular characteristics of E. gymnocyprii in four species of schizothoracine fishes from the QTP were explored.
Methods
We investigated the infection status of E. gymnocyprii in 156 schizothoracine fishes from the upper Yangtze River, upper Yellow River, and Qinghai Lake in Qinghai Province, China. The complete internal transcribed spacer (ITS) of the ribosomal RNA (rRNA) gene and part of the mitochondrial cytochrome c oxidase subunit 1 (cox1) gene of 35 E. gymnocyprii isolates from these fishes were sequenced and their characteristics analyzed. In addition, we inferred phylogenetic relationships of the E. gymnocyprii populations based on the rRNA-ITS and cox1 sequences.
Results
The total prevalence of E. gymnocyprii in schizothoracine fishes was 57.69% (90/156). However, the prevalence among different species as well as that across the geographical locations of the schizothoracine fishes was significantly different. The results of sequence analysis showed that the four E. gymnocyprii populations from different hosts and regions of Qinghai Province were conspecific, exhibiting rich genetic diversity. Phylogenetic analysis based on rRNA-ITS and cox1 sequences supported the coalescence of branches within E. gymnocyprii; the cox1 gene of E. gymnocyprii populations inferred some geographical associations with water systems. In addition, three species of schizothoracine fishes were recorded as new definitive hosts for E. gymnocyprii.
Conclusions
To the best of our knowledge, this is the first molecular description of E. gymnocyprii populations in schizothoracine fishes from the Qinghai-Tibet Plateau that provides basic data for epidemiological surveillance and control of acanthocephaliasis to protect endemic fish stocks.

Similar content being viewed by others

Background
Acanthocephalans are a group of widely distributed intestinal helminths, commonly found in fishes worldwide; they can cause malnutrition, tissue injury, and intestinal obstruction in hosts [1,2,3]. Echinorhynchus Zoega in Müller, 1776 (Acanthocephala: Echinorhynchidae) is a group of spiny-headed worms that is mainly parasitic in teleost fish and crustaceans found in various aquatic environments. It is the most widely distributed genus of acanthocephalans [4]; 52 species of Echinorhynchus have been recorded [5]. Because of its high morphological variability, there is a debate on the subdivision of this genus, and morphological identification of the species of Echinorhynchus is difficult [4, 6]. Therefore, DNA sequence data from molecular markers have been used for classification, evolution, and phylogeny of the Echinorhynchus spp. [4, 7, 8]. Echinorhynchus gymnocyprii Liu, Wang & Yang, 1981, with an elongated and cylindrical proboscis, and 14–16 longitudinal rows of hooks (each row having 10–11 hooks of unequal length), was first described in Gymnocypris przewalskii (Kessler, 1876) in the Qinghai Lake of China [9]. Later, its presence was discovered in Triplophysa scleroptera (Herzenstein, 1888) in the Qinghai Lake and in T. stenura (Herzenstein, 1888) in the Lhasa River in Tibet [10, 11]. However, to date, there has been no molecular data on E. gymnocyprii.
The schizothoracine fishes (Teleostei: Cyprinidae) are a dominant fish group of the Qinghai Tibet Plateau (QTP) ichthyofauna, and China has the largest distribution of schizothoracine species worldwide [12,13,14]. These fishes are endemic and adapted to the extreme environmental conditions (e.g. hypoxia, strong ultraviolet rays, food shortages and cold) of the QTP [12, 15,16,17]. Therefore, they are ideal models to investigate speciation, biological evolution, ecological effects, paleo-drainage changes, and biogeography of the QTP [13, 17,18,19,20]. However, a variety of factors have led to a sharp decline in the population of schizothoracine fishes in the last decades [21, 22]. Parasites found in these fishes may contribute to this decline, as some studies suggest that parasites may have adverse effects on their host populations [23, 24]. As an important part of QTP biodiversity, parasites of endemic fishes have gradually attracted the attention of researchers [11, 25].
During the investigation of the helminth fauna of schizothoracine fishes in the QTP of China, it was found that these fishes were infected with acanthocephalans, which were initially identified as E. gymnocyprii based on morphological studies. In the present study, the infection status of E. gymnocyprii in four species of schizothoracine fishes from the upper Yangtze River, Qinghai Lake, and upper Yellow River in the QTP was evaluated. The molecular data on E. gymnocyprii populations from different hosts and geographical locations (including the E. gymnocyprii population from the type-host, G. przewalskii, and type-locality of Qinghai Lake) are described based on ribosomal RNA (rRNA), which includes the complete internal transcribed spacer (ITS) and the mitochondrial cytochrome c oxidase subunit 1 (cox1) gene. In addition, the phylogenetic relationships of the E. gymnocyprii populations were investigated based on the rRNA-ITS and cox1 sequences.
Methods
Sampling
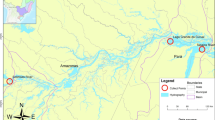
A total of 156 schizothoracine fishes (20 Ptychobarbus kaznakovi Nikolskii, 1903, 73 Gymnocypris eckloni Herzenstein, 1891, 11 Gymnodiptychus pachycheilus Herzenstein, 1892, and 52 Gymnocypris przewalskii), from four localities and three water systems in the northeastern QTP of Qinghai Province, China, were examined for parasites (Fig. 1). The sampling locations were in Zhiduo County (ZD) of the upper Yangtze River, Maduo County (MD) and Dari County (DR) of the upper Yellow River, and Qinghai Lake (QHL); the altitudes of ZD, MD, DR, and QHL are 4285 m, 4300 m, 3970 m and 3196 m, respectively. The Yangtze and Yellow rivers are both freshwater rivers, and the Qinghai Lake is a closed-basin brackish lake. The acanthocephalans were washed immediately in saline after collection from the intestines of fishes, preserved in 70% ethanol, and stored at 4 °C. The preliminary identification of E. gymnocyprii was mainly based on morphological characteristics (elongated and cylindrical proboscis, armed with 14–16 longitudinal rows of hooks, each row having 10–11 hooks of unequal length) [9, 26]. Subsequently, 35 acanthocephalans were selected for DNA analysis. The E. gymnocyprii found in G. przewalskii of Qinghai Lake was the type-species.

Collection sites of E. gymnocyprii in the Qinghai Province of China
DNA extraction, amplification, cloning and sequencing
Before performing genomic DNA analysis, the anterior extremities of the acanthocephalans were cut off to avoid contaminating the DNA with fish tissues attached to the proboscis. Next, the genomic DNA of individual acanthocephalans was extracted using the QIAamp® DNA Mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s recommendations.
We used the primers 18SF1 (5′-GCG AAG CAT TTG CCA AGA A-3′) and NC2 (5′-TTA GTT TCT TTT CCT CCG CT-3′) to amplify the fragment of rRNA-ITS (including partial 18S rRNA gene, ITS1; 5.8S rRNA gene, ITS2; and partial 28S rRNA gene) [7]. A partial DNA fragment of the cox1 gene was also amplified using the forward primer (5′-AGT TCT AAT CAT AA(R) GAT AT(Y) GG-3′) and the reverse primer (5′-TAA ACT TCA GGG TGA CCA AAA AAT CA-3′) [27].
Each PCR mixture (50 μl) consisted of 25 μl 2× pfu PCR MasterMix (Zomanbio, Beijing, China), 0.5 μl of each primer (0.5 μM), 23 μl ddH2O, and 1 μl genomic DNA. The PCR conditions for each gene were as follows: rRNA-ITS [7]: 93 °C for 3 min (pre-denaturation), followed by 35 cycles at 93 °C for 1 min (denaturation), 48 °C for 1 min (annealing), 72 °C for 2 min (extension), followed by a final extension step at 72 °C for 3 min; cox1 gene: 95 °C for 5 min (pre-denaturation), 94 °C for 30 s (denaturation), 52 °C for 30 s (annealing), 72 °C for 1 min (extension), each for 35 cycles, and a final extension step at 72 °C for 10 min. All amplicons were tested by electrophoresis on a 1% agarose gel with ExRed (Zomanbio). The PCR products were purified using a DNA Gel Extraction Kit (Axygen, Union, USA), cloned with a pMD™19-T vector, and then transformed in DH5α Escherichia coli cells (TaKaRa, Dalian, China). The positive plasmid DNA samples were sent to Sangon Company (Shanghai, China) for sequencing.
Sequence analysis
The sequence obtained from each acanthocephalan was assembled using DNAStar software [28] and then aligned with Clustul X 2.0 [29] and edited manually. Subsequently, the data obtained from these corrected sequences were analyzed with MEGA 6.06; both the pairwise distance and the mean distance within/between the populations were estimated using the Kimura 2-parameter model [30, 31]. In addition, the sequences were compared with sequences from GenBank by BLAST analysis. The sequences of ITS1 and ITS2 were defined in a previous study [32]. Based on the ITS1, ITS2 and cox1 sequences of each population, haplotype analysis was carried out using DNASP 5.10 [33].
Phylogenetic analysis
The rRNA-ITS and cox1 sequences were independently analyzed and aligned with sequences from GenBank. The rRNA-ITS sequences were as follows: E. gadi Zoega in Müller, 1776 (EF107647, EF107648), Pomphorhynchus laevis Zoega in Müller, 1776 (KJ756500), P. bosniacus Kistaroly & Cankovic, 1969 (MH319900), and P. zhoushanensis (KY472823); the cox1 sequences were: E. gadi (AY218095), E. salmonis Müller, 1784 (KP261017), E. truttae Schrank, 1788 (DQ089710), and Acanthocephalus lucii Müller, 1776 (KP261016), according to the software Clustal X 2.0 [29]. After amendment, the rRNA-ITS and cox1 datasets included 633 characters and 585 characters, respectively. Phylogenetic trees were constructed by the neighbor-joining (NJ) method using MEGA 6.06 [30] and Maximum Likelihood (ML) with IQ tree 1.6.10 [34]. For NJ, the Kimura 2-parameter model was used. The TPM3 + F + I for the rRNA-ITS dataset, and the HKY + F + I for the cox1 dataset, were suggested as the best-fit models by ModelFinder [35] of IQ tree 1.6.10 based on the Bayesian information criterion (BIC). Pomphorhynchus zhoushanensis and A. lucii were selected as the outgroups of the rRNA-ITS and cox1 datasets, respectively. The branch reliability of the phylogenetic tree constructed by the two methods was tested by bootstrapping with 1000 replicates. A haplotype network was constructed from the cox1 DNA sequence data with the software TCS using the probabilistic method of statistical parsimony [36, 37].
Statistical analysis
The differences in E. gymnocyprii infection rates among different fishes and locations were analyzed using the chi-square test in SPSS 25.0 for Windows (IBM Corp., New York, USA). P < 0.05 was considered as statistically significant.
Results
Prevalence of E. gymnocyprii in schizothoracine fishes
The morphological characters of E. gymnocyprii based on samples collected from different hosts and locations were similar, with a minor variation in size (Additional file 1: Table S1). Echinorhynchus gymnocyprii was found in 90 of 156 (57.69%) fish samples, with infection rates ranging from 45% (G. pachycheilus) to 100% (P. kaznakovi) among the four species of schizothoracine fishes (χ2 = 15.919, df = 3, P = 0.0012), The prevalence of these helminths varied across different geographical localities; among the four locations in Qinghai Province, the prevalence ranged from 40.00% in Dari to 100.00% in Maduo (χ2 = 26.425, df = 3, P < 0.0001) (Table 1). In addition, the mean intensity of E. gymnocyprii infection ranged from 14.9 (G. eckloni in Dari) to 41.4 (G. pachycheilus in Maduo) (Table 1).
Sequence analysis
The locations, sample numbers, sex, and hosts of E. gymnocyprii are shown in Table 2. The rRNA-ITS region was 1493-1497 bp in length (259–263 bp for ITS1, 85 bp for 5.8S and 251 bp for ITS2 rDNA) (GenBank: MT162052–MT162085). The percentages of nucleotide identity and variations in the number of nucleotides within E. gymnocyprii populations, based on ITS1 (259–263 bp) and ITS2 (251 bp), are shown in Additional file 2: Table S2 and Additional file 3: Table S3, respectively. The ITS1 of E. gymnocyprii from QHL4-5, MD1-3, MD7-13, MD15-17 and DR2-5 were 100% identical; ZD1, ZD3 and ZD5 were 100% identical (where QHL: Qinghai Lake; MD: Maduo; DR: Dari; and ZD: Zhiduo), and the percent identity within E. gymnocyprii was 96.9–100.0%. The ITS2 of QHL1, QHL3-5, MD2, MD4-7, MD9-13, MD15-18, DR2-4 and ZD1-5 were 100% identical, despite being found at different locations (QHL, MD, DR and ZD) and in different hosts (G. przewalskii, G. eckloni, Gymnodiptychus pachycheilus and P. kaznakovi); the percent identity within E. gymnocyprii was 98.4–100%. The percent identity based on the ITS1 and ITS2 between E. gymnocyprii and E. gadi (GenBank: EF107647) was 54.0–56.2% (Additional file 2: Table S2, Additional file 3: Table S3).
Regarding the cox1 gene, 35 newly generated sequences were 702 bp long; the sequences of QH1 and QH5 were identical, and the sequences of ZD4 and ZD5 were identical. The overall cox1 GC content was 37.3%, showing a high AT bias; the percent identity between these sequences was 95.4–100.0%. The overall average of pairwise distances was 0.02182, and the percent identity/mean distance within each population was 98.8–100.0%/0.00599 for MD, 99.0–99.7%/0.00697 for DR, 98.8–100.0%/0.00862 for QHL, and 99.3–100.0%/0.00401 of ZD, respectively. The percent identity/mean distance of the QHL population and MD, DR and ZD populations was 95.9–97.3%/0.0344, 96.8–98.1%/0.0236 and 95.4–96.6%/0.0385, respectively. There are six other Echinorhynchus species with cox1 gene sequences deposited on GenBank, and the mean pairwise comparison between the E. gymnocyprii population in our study and these species yielded 0.2386 (E. salmonis, GenBank: KP261017) to 0.4263 (E. cinctulus, GenBank: KP261014) nucleotide differences. The cox1 sequences of E. gymnocyprii have been deposited in GenBank under the accession numbers MT169741–MT169775. The summary statistics for the cox1 gene in E. gymnocyprii populations are shown in Table 3, and the values of h, k, and π indicate that the populations of E. gymnocyprii are rich in genetic diversity.
Phylogenetic analyses
The topological structures of the phylogenetic trees based on the rRNA-ITS and cox1 sequences of the E. gymnocyprii populations were similar; all the E. gymnocyprii populations clustered in a single well-supported clade, with only slight differences in the bootstrap values for some nodes (Figs. 2, 3, 4). The phylogenetic NJ tree of E. gymnocyprii populations based on the cox1 sequences, inferred 4 subclades (subclades MD, DR, QHL and ZD; Fig. 4), corresponding to 4 geographical locations belonging to 3 water systems. The MD and DR groups belong to the Yellow River system, the QHL group belongs to the Qinghai Lake system, and the ZD group belongs to the Yangtze River system. However, the DR and ZD groups appear to be paraphyletic in the ML tree based on the cox1 sequences (Fig. 3).

Phylogenetic relationships of E. gymnocyprii. The tree is inferred from the rRNA-ITS sequences using neighbor-joining (NJ) method. The maximum likelihood (ML) method produced phylogenetic tree with the same branch topologies. Bootstrap support from ML/NJ analysis are shown above the nodes. The scale-bar indicates the number of substitutions per site. Pomphorhynchus zhoushanensis was used as the outgroup

Phylogenic tree based on cox1 gene sequences of E. gymnocyprii using the maximum likelihood (ML) method. Bootstrap support is shown above the nodes. The scale-bar indicates the number of substitutions per site. Branch labels of different colors indicate different locations of E. gymnocyprii populations in this study (blue, Maduo County (MD); red, Dari County (DR); purple, Qinghai Lake (QHL); green, Zhiduo County (ZD).) Acanthocephalus lucii was used as the outgroup. The branches of the phylogram depicting the relationships among the E. gymnocyprii sequences are shown in Additional file 4: Figure S1

Phylogenic tree based on cox1 gene sequences of E. gymnocyprii using the Neighbor-Joining (NJ) method. Bootstrap support is shown above the nodes. The scale-bar indicates genetic distance. Branch labels of different colors indicate different sources of E. gymnocyprii populations in this study (blue, Maduo County (MD); red, Dari County (DR); purple, Qinghai Lake (QHL); green, Zhiduo County (ZD)). Acanthocephalus lucii was used as the outgroup
The 35 specimens comprised 33 unique haplotypes, and the statistical parsimony analysis revealed 3 distinct networks (QHL, ZD and MD-DR; Fig 5), corresponding to three water systems. Network QHL contained 4 haplotypes from 5 individuals, QHL1 and QHL5 were considered the central haplotype; network ZD contained 4 haplotypes from 5 individuals, ZD4 and ZD5 were considered the central haplotype; network MD-DR contained 25 haplotypes from 25 individuals, with MD13 as the ancestral haplotype.

Statistical parsimony network of E. gymnocyprii haplotype based on cox1 gene sequences. The connection limit excluding homoplasic changes was set to 95%. Each oval represents a haplotype, and the ancestral haplotype (with the highest outgroup probability) is indicated by a square, the size of the square or oval corresponds the haplotype frequency. Each line equates to one mutational step, and the small circles are hypothetical haplotypes
Discussion
In this study, we found that the prevalence and intensity of E. gymnocyprii from G. przewalskii in Qinghai Lake had similarity to those reported in previous studies [10]. Previously, a range of 33.9–100% for prevalence and 14.9–41.4 for mean intensity of E. gymnocyprii in G. eckloni, Gymnodiptychus pachycheilus, and P. kaznakovi was reported. In addition, the prevalence of E. gymnocyprii in schizothoracine fishes differed significantly depending on fish species and location. There are several possible explanations for these differences, such as the characteristics (e.g. feeding behavior, age, physiology and abundance) of different definitive hosts, different numbers of samples, and the ecological environment (e.g. density of intermediate hosts, aquatic organisms, water conditions and altitude) [38,39,40,41,42,43,44]; these differences result in different survival conditions for both the schizothoracine fishes and acanthocephalans. Our results suggest that E. gymnocyprii is widely distributed in native fishes of the QTP, a finding that should be considered when working to protect the schizothoracine fish population. Further studies of E. gymnocyprii may serve as a model for studying the co-evolution of native fishes and parasites in the QTP.
However, the host species, geographical distribution, age, and other factors may cause changes in the morphology of Echinorhynchus spp. These changes sometimes exceed the suggested generic boundaries, thereby leading to inconvenience and confusion in the classification and identification of Echinorhynchus spp. [2, 4, 6, 45,46,47]. Therefore, DNA sequences from the ribosomal gene cluster (e.g. 18S, ITS and 28S) or mitochondria (e.g. cox1) provide a complementary methodology for analyzing the taxonomy and phylogeny of Echinorhynchus spp. [4, 7, 47]. Taken together, these results support the use of rRNA (containing ITS) and the cox1 gene as genetic markers for the identification of E. gymnocyprii.
In our study, the values of π, k, and Hd based on the cox1 gene indicated that the E. gymnocyprii populations exhibit rich genetic diversity [33], which may explain the existence of these populations in a variety of endemic fish in the QTP and their adaptation to the harsh environment of the plateau. The overall π (0.02122) and k (14.95966) were significantly higher than those of π (0.00397–0.00851) and k (2.800–6.000) of single populations from MD, DR, QHL and ZD, which indicates that geographical factors may have a positive impact on the evolution of the cox1 gene in the E. gymnocyprii populations.
The phylogenetic analysis based on the rRNA-ITS and cox1 gene supports the coalescence of branches within the E. gymnocyprii populations, and provides evidence that these populations belong to the same species. Interestingly, the NJ tree based on the cox1 gene showed four subclades (Fig. 4), and the statistical parsimony analysis revealed three distinct networks (Fig. 5), both reflecting geographical associations with water systems. The E. gymnocyprii populations from the Yellow River system (MD, DR and QHL) were more closely related to each other than to those of the Yangtze River system (ZD). This result might be explained by geological research findings, which imply that the Qinghai Lake was connected to the ancient Yellow River approximately 0.15 mega-annum before the present (Ma BP) [48]. The distribution and evolution of schizothoracine fishes, as hosts of E. gymnocyprii, in the QTP are strictly due to differences in water systems and habitats [15, 49,50,51,52]. The cox1 gene of E. gymnocyprii populations showed some geographical associations with water systems, indicating that adaptive changes in the cox1 gene enabled the species to survive in different water environments and preserve genetic differentiation. Consequently, the cox1 gene is potentially useful for investigating the zoogeography of E. gymnocyprii.
In the present study, to the best of our knowledge, two DNA markers of E. gymnocyprii were analyzed for the first time, and three new host records were added, which may be the first step in further understanding the speciation and evolution of this species in the QTP. This study also provides important molecular data for revision of the genus Echinorhynchus [4] in the future.
Conclusions
Echinorhynchus gymnocyprii is widely distributed in native fish of QTP; three species of schizothoracine fishes (G. eckloni, Gymnodiptychus pachycheilus and P. kaznakovi) were identified as new definitive hosts of E. gymnocyprii. Our study is the first molecular characterization of E. gymnocyprii populations in schizothoracine fishes from the QTP; it provides basic data for epidemiological surveillance and control of acanthocephaliasis to protect endemic fish stocks in the QTP.
Availability of data and materials
The datasets supporting the findings of this article are included within the article and its additional files. The nuclear rDNA-ITS and cox1 nucleotide sequences generated in this study were deposited in the GenBank database under the accession numbers MT162052–MT162085 and MT169741–MT169775, respectively.
Abbreviations
- BIC:
-
Bayesian information criterion
- C:
-
conserved sites
- ITS:
-
internal transcribed spacer
- k:
-
average number of nucleotide difference
- Ma BP:
-
mega-annum before present
- ML:
-
maximum likelihood
- NJ:
-
neighbor-joining
- Pi:
-
parsimony-informative sites
- QTP:
-
Qinghai-Tibetan plateau
- S:
-
singleton sites
- V:
-
variable sites
References
Nabi S, Tanveer S, Ganaie SA, Niyaz U, Abdullah I. Acanthocephalan infestation in fishes—a review. J Zool St. 2015;2:33–8.
Sheema SH, John MV, George PV. SEM studies on acanthocephalan parasite, Echinorhynchus veli infecting the fish Synaptura orientalis (Bl & Sch, 1801). J Parasit Dis. 2016;41:71–5.
Sanil NK, Asokan PK, John L, Vijayan KK. Pathological manifestations of the acanthocephalan parasite, Tenuiproboscis sp. in the mangrove red snapper (Lutjanus argentimaculatus) (Forsskål, 1775), a candidate species for aquaculture from southern India. Aquaculture. 2011;310:259–66.
Wayland MT, Vainio JK, Gibson DI, Herniou EA, Littlewood DT, Vainola R. The systematics of Echinorhynchus Zoega in Muller, 1776 (Acanthocephala, Echinorhynchidae) elucidated by nuclear and mitochondrial sequence data from eight European taxa. ZooKeys. 2015;484:25–52.
Amin OM. Classification of the Acanthocephala. Folia Parasitol. 2013;60:273–305.
Shostak AW, Dick TA, Szalai AJ, Bernier LM. Morphological variability in Echinorhynchus gadi, E. leidyi, and E. salmonis (Acanthocephala: Echinorhynchidae) from fishes in northern Canadian waters. Can J Zool. 1986;64:985–95.
Sobecka E, Szostakowska B, MacKenzie K, Hemmingsen W, Prajsnar S, Eydal M. Genetic and morphological variation in Echinorhynchus gadi Zoega in Muller, 1776 (Acanthocephala: Echinorhynchidae) from Atlantic cod Gadus morhua L. J Helminthol. 2012;86:16–25.
Weber M, Wey-Fabrizius AR, Podsiadlowski L, Witek A, Schill RO, Sugar L, et al. Phylogenetic analyses of endoparasitic Acanthocephala based on mitochondrial genomes suggest secondary loss of sensory organs. Mol Phylogenet Evol. 2013;66:182–9.
Liu LQ, Wang BZ, Yang T. Study on the Acanthocephalans in the Gymnocypris przewalskii przewalskii in the Qinghai Lake. J Fish China. 1981;5:295-9+373-5 (in Chinese).
Yang TB, Liao XH. Seasonal population dynamics of Echinorhynchus gymnocyprii (Acanthocephala: Echinorhynchidae) in the host Gymnocypris przaewalskii przewalskii in the Qinghai Lake. Acta Sci Nat Univ Sunyatseni. 2000;39:78–82.
Li WX, Zhang LX, Gao Q, Nie P. Endohelminths and their community characteristics in fish of the Lhasa River in Tibet of China. Chin J Zool. 2008;43:1–8.
Liang Y, He D, Jia Y, Sun H, Chen Y. Phylogeographic studies of schizothoracine fishes on the central Qinghai-Tibet Plateau reveal the highest known glacial microrefugia. Sci Rep. 2017;7:10983.
Tang Y, Li C, Wanghe K, Feng C, Tong C, Tian F, et al. Convergent evolution misled taxonomy in schizothoracine fishes (Cypriniformes: Cyprinidae). Mol Phylogenet Evol. 2019;134:323–37.
Wu YF, Wu CZ. Fishes of the Qinghai-Xizang plateau. Chendu: Sichuan Publishing House of Science & Technology; 1992.
Duan Z, Zhao K, Peng Z, Li J, Diogo R, Zhao X, et al. Comparative phylogeography of the Yellow River schizothoracine fishes (Cyprinidae): vicariance, expansion, and recent coalescence in response to the Quaternary environmental upheaval in the Tibetan Plateau. Mol Phylogenet Evol. 2009;53:1025–31.
He D, Chen Y. Biogeography and molecular phylogeny of the genus Schizothorax (Teleostei: Cyprinidae) in China inferred from cytochrome b sequences. J Biogeogr. 2006;33:1448–60.
Tong C, Fei T, Zhang C, Zhao K. Comprehensive transcriptomic analysis of Tibetan Schizothoracinae fish Gymnocypris przewalskii reveals how it adapts to a high altitude aquatic life. BMC Evol Biol. 2017;17:74.
Tong C, Tian F, Zhang C, Zhao K. The microRNA repertoire of Tibetan naked carp Gymnocypris przewalskii: a case study in Schizothoracinae fish on the Tibetan Plateau. PLoS ONE. 2017;12:e0174534.
Qi D, Guo S, Chao Y, Kong Q, Li C, Xia M, et al. The biogeography and phylogeny of schizothoracine fishes (Schizopygopsis) in the Qinghai-Tibetan Plateau. Zool Scr. 2015;44:523–33.
Zhang R, Peng Z, Li G, Zhang C, Tang Y, Gan X, et al. Ongoing speciation in the Tibetan Plateau Gymnocypris species complex. PLoS ONE. 2013;8:e71331.
Liu G, Wu Y, Shen X, Hu Y, Qin X, Tian W, et al. Laboratory experiments demonstrate that the hissing of the Chinese alligator can effectively inhibit movement of flower fish Ptychobarbus kaznakovi. Hydrobiologia. 2019;836:97–108.
Su J, Ji W, Zhang Y, Gleeson DM, Lou Z, Ren J, et al. Genetic diversity and demographic history of the endangered and endemic fish (Platypharodon extremus): implications for stock enhancement in Qinghai Tibetan Plateau. Biol Fishes. 2015;98:763–74.
Esch GW, Fernández JC. Influence of parasites on host populations. In: Esch GW, Fernández JC, editors. A functional biology of parasitism: ecological and evolutionary implications. Dordrecht: Springer; 1993. p. 91–119.
Lafferty KD. Ecosystem consequences of fish parasites. J Fish Biol. 2008;73:2083–93.
Xi BW, Oros M, Chen K, Xie J. A new monozoic tapeworm, Parabreviscolex niepining, n. sp. (Cestoda: Caryophyllidea), from schizothoracine fishes (Cyprinidae: Schizothoracinae) in Tibet. China. Parasitol Res. 2018;117:347–54.
Amin OM. Classification. In: Crompton DWT, Nickol BB, editors. Biology of the Acanthocephala. Cambridge: Cambridge University Press; 1980. p. 27–72.
Garcia-Varela M, Perez-Ponce de Leon G, Aznar FJ, Nadler SA. Phylogenetic relationship among genera of Polymorphidae (Acanthocephala), inferred from nuclear and mitochondrial gene sequences. Mol Phylogenet Evol. 2013;68:176–84.
Burland TG. DNASTAR’s Lasergene sequence analysis software. In: Misenser S, Krawetz SA, editors. Bioinformatics methods and protocol. Totowa: Springer; 2000. p. 71–91.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–82.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–20.
Král’ová-Hromadová I, Tietz DF, Shinn AP, Špakulová MITS. ITS rDNA sequences of Pomphorhynchus laevis (Zoega in Müller, 1776) and P. lucyi Williams & Rogers, 1984 (Acanthocephala: Palaeacanthocephala). Syst Parasitol. 1984;2003(56):141–5.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Kalyaanamoorthy S, Minh BQ, Wong TK, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587.
Templeton AR, Crandall K, Sing CF. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics. 1992;132:619–33.
Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–9.
Song Z, Proctor H. Parasite prevalence in intermediate hosts increases with waterbody age and abundance of final hosts. Oecologia. 2020;192:311–21.
Atalabi TE, Awharitoma AO, Akinluyi FO. Prevalence, intensity, and exposed variables of infection with Acanthocephala parasites of the gastrointestinal tract of Coptodon zillii (Gervais, 1848) (Perciformes: Cichlidae) in Zobe Dam, Dutsin-Ma Local Government Area, Katsina State, Nigeria. J Basic Appl Zool. 2018;79:29.
Sakthivel A, Selvakumar P, Gopalakrishnan A. Acanthocephala (Acanthocephalus lucii) infection on Caranx ignobilis from Nagapattinam, south east coast of India. Indian J Geo-Mar Sci. 2016;45:448–52.
Nachev M, Sures B. Seasonal profile of metal accumulation in the acanthocephalan Pomphorhynchus laevis: a valuable tool to study infection dynamics and implications for metal monitoring. Parasit Vectors. 2016;9:300.
Lagrue C, Poulin R. Bottom-up regulation of parasite population densities in freshwater ecosystems. Oikos. 2015;124:1639–47.
Aydogdu A, Emre N, Emre Y. Prevalence and intensity of parasitic helminths of thicklip grey mullet Chelon labrosus in hosts in Beymelek Lagoon Lake in Antalya, Turkey, according to season, host size, age, and sex of the host. Turk J Zool. 2015;39:643–51.
Zdzitowiecki K, Laskowski Z. New data on the occurrence of Acanthocephala in some fish in Admiralty Bay (South Shetland Islands). Acta Parasitol. 2013;58:547–50.
Amin OM, Heckmann RA, Radwan NAE, Anchundia JSM, Alcivar MAZ. Redescription of Rhadinorhynchus ornatus (Acanthocephala: Rhadinorhynchidae) from skipjack tuna, Katsuwonus pelamis, collected in the Pacific Ocean off South America, with special reference to new morphological features. J Parasitol. 2009;95:656–64.
Amin OM, Redlin MJ. The effect of host species on growth and variability of Echinorhynchus salmonis Müller, 1784 (Acanthocephala: Echinorhynchidae), with special reference to the status of the genus. Syst Parasitol. 1980;2:9–20.
Väinölä R, Valtonen E, Gibson D. Molecular systematics in the acanthocephalan genus Echinorhynchus (sensu lato) in northern Europe. Parasitology. 1994;108:105–14.
Zhang Z, Yu Q, Zhang K, Gu Y, Xing S. Geomorphological evolution of Quaternary River from upper Yellow River and geomorphological evolution investigation for 1:250000 scale geological mapping in Qinghai-Tibet plateau. J Earth Sci. 2003;28:621–6.
Zhao K, Li J, Yang G, Duan Z, He S, Chen Y. Molecular phylogenetics of Gymnocypris (Teleostei: Cyprinidae) in Lake Qinghai and adjacent drainages. Chin Sci Bull. 2005;50:1326–34.
Zhao K, Duan ZY, Peng ZG, Guo SC, Li JB, He SP, et al. The youngest split in sympatric schizothoracine fish (Cyprinidae) is shaped by ecological adaptations in a Tibetan Plateau glacier lake. Mol Ecol. 2009;18(17):3616–28.
He D, Chen Y, Chen Y, Chen Z. Molecular phylogeny of the specialized schizothoracine fishes (Teleostei: Cyprinidae), with their implications for the uplift of the Qinghai-Tibetan Plateau. Chin Sci Bull. 2004;49:39–48.
He D, Chen Y. Molecular phylogeny and biogeography of the highly specialized grade schizothoracine fishes (Teleostei: Cyprinidae) inferred from cytochrome b sequences. Chin Sci Bull. 2007;52(6):777–88.
Acknowledgements
The authors would like to thank all the people who were involved in making this project a success.
Funding
This study was supported by the International Scientific and Technological Cooperation project of Qinghai Province (Grant No. 2010-H-810).
Author information
Authors and Affiliations
Contributions
YMZ and JZC conceived and designed the study. MTL performed the experiments, analyzed the data and drafted the manuscript. CHL, JS and DDM collected the samples, YF modified the manuscript. YPL identified the fish samples. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Research Ethics Committee of Northwest A&F University. All fish samples were collected after the permission of the Qinghai Fishery Administration Station, and all procedures is performed strictly in accordance with the requirements of Animal Ethics Procedures and Guidelines of the People’s Republic of China.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Comparison of the specific characters of E. gymnocyprii from different hosts and locations.
Additional file 2: Table S2.
Percentage nucleotide identity and numbers of nucleotide variations of ITS1 fragments from E. gymnocyprii populations.
Additional file 3: Table S3.
Percentage nucleotide identity and numbers of nucleotide variations of ITS2 fragments from E. gymnocyprii populations.
Additional file 4: Figure S1.
A phylogram form maximum likelihood (ML) analysis based on cox1 gene sequences depicting the relationships among the E. gymnocyprii sequences.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lei, MT., Cai, JZ., Li, CH. et al. Prevalence and genetic diversity of Echinorhynchus gymnocyprii (Acanthocephala: Echinorhynchidae) in schizothoracine fishes (Cyprinidae: Schizothoracinae) in Qinghai-Tibetan Plateau, China. Parasites Vectors 13, 357 (2020). https://doi.org/10.1186/s13071-020-04224-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-020-04224-w