
Abstract
Background
Trypanosoma cruzi, the causative agent of Chagas disease, and T. rangeli are kinetoplastid parasites endemic to Latin America. Although closely related to T. cruzi and capable of infecting humans, T. rangeli is non-pathogenic. Both parasite species are transmitted by triatomine bugs, and the presence of T. rangeli constitutes a confounding factor in the study of Chagas disease prevalence and transmission dynamics. Trypanosoma cruzi possesses high molecular heterogeneity: seven discrete typing units (DTUs) are currently recognized. In Ecuador, T. cruzi TcI and T. rangeli KP1(-) predominate, while other genetic lineages are seldom reported.
Methods
Infection by T. cruzi and/or T. rangeli in different developmental stages of triatomine bugs from two communities of southern Ecuador was evaluated via polymerase chain reaction product size polymorphism of kinetoplast minicircle sequences and the non-transcribed spacer region of the mini-exon gene (n = 48). Forty-three mini-exon amplicons were also deep sequenced to analyze single-nucleotide polymorphisms within single and mixed infections. Mini-exon products from ten monoclonal reference strains were included as controls.
Results
Trypanosoma cruzi genetic richness and diversity was not significantly greater in adult vectors than in nymphal stages III and V. In contrast, instar V individuals showed significantly higher T. rangeli richness when compared with other developmental stages. Among infected triatomines, deep sequencing revealed one T. rangeli infection (3%), 8 T. cruzi infections (23.5%) and 25 T. cruzi + T. rangeli co-infections (73.5%), suggesting that T. rangeli prevalence has been largely underestimated in the region. Furthermore, deep sequencing detected TcIV sequences in nine samples; this DTU had not previously been reported in Loja Province.
Conclusions
Our data indicate that deep sequencing allows for better parasite identification/typing than amplicon size analysis alone for mixed infections containing both T. cruzi and T. rangeli, or when multiple T. cruzi DTUs are present. Additionally, our analysis showed extensive overlap among the parasite populations present in the two studied localities (c.28 km apart), suggesting active parasite dispersal over the study area. Our results highlight the value of amplicon sequencing methodologies to clarify the population dynamics of kinetoplastid parasites in endemic regions and inform control campaigns in southern Ecuador.

Similar content being viewed by others


Background
Chagas disease (CD) is caused by the kinetoplastid hemoflagellate parasite Trypanosoma cruzi and affects 6–7 million people worldwide [1]. This neglected disease is endemic to Latin America, where it poses risk of infection for > 65 million people and kills an estimated 50,000 every year [2]. Transmission in endemic countries primarily occurs via contact with the feces of an infected triatomine bug. However, other secondary mechanisms of transmission, such as blood transfusion, organ transplants, congenital transmission and laboratory accidents exist, and can cause infections in non-endemic regions where vectors are not present [3].
Trypanosoma cruzi has high molecular and phenotypic heterogeneity, and the existence of genetic lineages has been long recognized. Initially, three “zymodemes” (termed Z1, Z2 and Z3) were identified via multilocus enzyme electrophoresis [4, 5]. Subsequently, a wide variety of molecular markers have been developed and employed to demonstrate the existence of seven lineages or discrete typing units (DTUs), termed TcI-TcVI and Tcbat [6,7,8,9,10,11,12]. In Ecuador, TcI predominates in the central coast [13, 14] and southern highlands (Loja Province) [15]. Only two previous reports suggest the occurrence of parasites belonging to DTUs other than TcI, in triatomines and in patients of CD [16, 17].
In Ecuador, Triatoma dimidiata, T. carrioni, Panstrongylus chinai, P. rufotuberculatus and Rhodnius ecuadoriensis are epidemiologically relevant vector species [18, 19]. Rhodnius ecuadoriensis is widely distributed in the western lowlands and the southern provinces of the country, as well as in northern Peru [20], both in domestic and in peridomestic environments where contact with humans occurs [19, 21].
Trypanosoma rangeli, a related kinetoplastid parasite whose geographical distribution overlaps with that of T. cruzi, may also be found in the same triatomine vectors and mammalian hosts. Mixed infections with both species are often reported [18, 22, 23]. Despite being considered non-pathogenic to humans, T. rangeli is epidemiologically relevant since it may cause false-positive results in microscopical and serological tests used for diagnosis of T. cruzi infection [24, 25]. In contrast to that of T. cruzi, transmission of T. rangeli to vertebrate hosts occurs via the salivary route, although the possibility of transmission through infected feces has also been suggested [26, 27]. Upon ingestion from the circulation of vertebrate hosts, T. rangeli trypomastigotes accumulate in the digestive tract of triatomines, replicate, transform into epimastigotes, cross the intestinal epithelium into the hemocoel and migrate to the salivary glands. Infective metacyclic trypomastigotes are released with the saliva during a blood meal [28]. Little is known regarding T. rangeli in Ecuador. PCR minicircle amplification of > 3600 samples revealed that 10% of triatomines and mammals from Manabí and Loja provinces were infected with T. rangeli, while 1.25% presented T. cruzi + T. rangeli co-infections [29].
The kinetoplast DNA of trypanosomatid parasites is composed of maxicircles and minicircles, whose number varies between species. Minicircles possess a conserved 100–200 bp repetitive region, whose number of repeats also differs among species. In T. cruzi, there are four copies of the conserved sequence [30]. Trypanosoma rangeli may present one, two or four copies of the mini-repeat. These three different minicircle classes in T. rangeli are known respectively as KP1, KP2 and KP3 [31, 32]. Further analysis based on polymorphism of randomly amplified DNA (RAPD), sequencing of the small subunit of ribosomal RNA (SSU rRNA), the internal transcribed spacer of rDNA (ITS rDNA) and the intergenic region of the splice leader, has unveiled a more complex population structure for T. rangeli, with five groups defined as A, B, C, D and E, each of which shows a strong association with vector species and geographical distribution [27, 33, 34]. More recently, through the analysis of microsatellites and single-nucleotide polymorphisms (SNPs) of the splice leader gene, Sincero et al. [35] suggested the presence of three main groups in T. rangeli: (i) the Amazonian group, associated to Rhodnius brethesi and vertebrate hosts; (ii) the KP1(-) group, linked to the R. pallescens complex; and (iii) the KP1(+) group, related to the R. robustus complex.
Infection by T. cruzi in mammalian reservoirs and invertebrate vectors is frequently multiclonal, involving several intra-specific genotypes with genetically dissimilar profiles [36,37,38,39]. Multiclonal parasitic infections have been suggested to impact host immunity [40], disease transmission rate and population structure [41]. They can also mislead drug resistance evaluation, diagnostics and various other applications important to disease control [42].
In the genus Trypanosoma, mini-exon genes are present in several tandemly arranged copies and encode the splice leader, a 35-nucleotide sequence translocated to the 5’-end of every newly synthesized mRNA [43, 44] in a process referred to as discontinuous transcription [45]. Trypanosoma cruzi mini-exon genes include conserved, semi-conserved and highly variable regions, which have been used for phylogenetic analysis [46], discrimination between DTUs [47], population genetic inference [48], and more recently, in diversity analysis within naturally infected mammalian hosts [49]. Nonetheless, it is not clear how mini-exon sequence diversity relates to DTU identity, and restriction fragment length polymorphism (PCR-RFLP) assays targeting the mini-exon locus are not considered reliable for T. cruzi DTU detection and identification [50]. Given its variability, however, sequencing of the intergenic spacer has proven useful in the examination of population dynamics within a single DTU [50,51,52,53,54,55].
Next-generation sequencing (NGS) has helped provide insights into multiclonal T. cruzi infections in humans [56] and to assess relationships between gut microbiota, parasite diversity, and vertebrate feeding sources of triatomine bugs [57]. In the present study, we aimed to evaluate the molecular diversity of T. cruzi and T. rangeli in two localities of southern Ecuador via analysis of the mini-exon gene. We analyzed 48 intestinal DNA extracts from triatomines collected in Loja Province as well as DNA from ten cloned isolates of reference strain DTUs I, III, IV, V and VI in order to identify relationships between mini-exon diversity and DTU identity. Mini-exon amplicon sequence reads generated by NGS were sorted into haplotype clusters to measure genetic richness, diversity, variability and substructure in the landscape. Moreover, the dataset included samples isolated from different developmental stages of the vector to evaluate a potential correlation with mini-exon diversity. We also compared genotyping results from NGS and PCR product size polymorphism analysis in terms of sensitivity for discrimination between T. cruzi and T. rangeli parasites.
Methods
Sample panel and PCR typing
Archived DNA samples collected between years 2009–2013 by the Center for Research on Health in Latin America (CISeAL) were employed in the study. Details of entomological searches, intestinal content DNA extractions and corresponding approved protocols and collection permits have been reported elsewhere [19, 23]. Samples selected for the study had been previously genotyped by PCR amplification using primers 121 and 122, which anneal to highly conserved regions of the kinetoplast minicircle [58] and are useful to distinguish between T. cruzi (330 bp fragment size expected) and T. rangeli (380 bp). All samples appeared to be infected exclusively with T. cruzi based on visualization of 121/122 amplicons (data not shown).
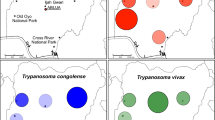
Forty-eight samples from two rural communities from southern Ecuador (Loja Province) were included in the sample panel: 31 from Bramaderos (4°4′47″S, 79°49′28″W); and 17 from Bellamaría (4°12′41″S, 79°36′23″W) (Fig. 1). Hypothesizing that infection multiclonality might accumulate with blood meal number, we characterized the molecular diversity/multiclonality of the mini-exon gene of the parasites across vector developmental stages, including samples obtained from 20 adult, 14 instar V, 3 instar IV and 11 instar III triatomine bugs.

Study area. The two studied communities within Loja Province in southern Ecuador are depicted. They are separated by approximately 28.3 km. Circle diameter is proportional to sample size in each locality. Map generated with ArcGIS software, version 10.5, based on data freely available from the Military Geographic Institute of Ecuador (IGM, http://www.geoportaligm.gob.ec/portal/). Community location was obtained via GPS measurement
The mini-exon gene is a multicopy target. DNA from 10 biologically cloned reference strains was therefore included in the analysis to evaluate whether intra-clonal sequence diversity generated from the amplicons via NGS recapitulated the DTU to which the clones had been previously assigned via classical methods [50].
Multiplex PCR
PCR amplification of the non-transcribed spacer region of the mini-exon gene was performed employing five primers in a multiplex reaction to discriminate between zymodemes I, II, III and T. rangeli, as previously described [47]. In the current nomenclature zymodeme I is equivalent to DTU TcI, zymodeme II comprises DTUs II, V and VI, while zymodeme III corresponds to DTUs III and IV. The amplified products were electrophoresed in 2% agarose gels and visualized with SYBR green under UV light. Samples co-infected with T. cruzi and T. rangeli displayed a two-band pattern. In such cases, gel excision and purification were performed for each fragment. Subsequently, amplicons were sequenced by NGS technology as detailed below.
Next-generation sequencing and bioinformatic analysis
Paired-end sequence reads were generated on the Illumina MiSeq platform (600-cycle Reagent Kit v3; Illumina, San Diego, USA) following a custom protocol [59]. Paired-end sequence reads were processed with Cutadapt v1.12 for adapter removal and Sickle v1.33 for quality trimming (-q 27) [60]. Trimmed forward and reverse reads were overlapped using default settings in Pandaseq v2.7 [61]. Eleven of 56 sample barcodes used during multiplexed sequencing in this study were also used for a separate study on 18S gene amplicons. These 18S amplicons were sequenced in the same MiSeq flow cell in order to save sequencing costs. We discarded all sequences containing 18S k-mer matches after the Pandaseq overlap step. Sequences with > 98% sequence similarity were then clustered to consensus haplotypes based on the UPARSE-OTU algorithm implemented by the ‘cluster_otus’ command in USEARCH v8.1 [62]. Chimeric sequences were discarded using the UCHIME algorithm and clusters represented by < 200 reads were discarded to avoid artefactual SNPs. Samples for which the ‘usearch_global’ command assigned < 20,000 reads to the remaining set of consensus sequences were also discarded based on rarefaction curves computed from cluster size annotations using ‘fasta_rarify’. Finally, parasite species and DTUs were determined for each haplotype by searching the complete nucleotide collection (nr/nt) with the Basic Local Alignment Search Tool (BLAST) at the National Center for Biotechnology Information (NCBI). All sequences with ≥ 90% similarity to trypanosomatid mini-exon sequences were kept for further analysis. (Additional file 1: Table S1 shows raw data for cloned reference strains and Ecuadorian samples).
Richness and diversity analysis
Richness was defined as the total number of haplotypes found in each sample. Shannonʼs index was defined as -Ʃ(Piln(Pi)) [63]. As mentioned previously, samples with mixed infections showed a two-band pattern in agarose gels. In these cases, gel excision rendered two sequencing templates and the information obtained was merged (addition of reads from each template) (Additional file 2: Table S2). For each sample, richness and diversity were calculated for both parasite species (T. cruzi and T. rangeli) independently and combined. Data corresponding to samples TBM2823, TBR1487 and TBR1489 were excluded from this analysis because they are the only representatives of instar IV. TBR1446 and TBR1503 were also excluded to avoid bias because the sequence reads from these samples has been merged across four sequencing reactions.
Statistical analysis
An analysis of variance (ANOVA) was performed when data showed normality and homoscedasticity. When data violated normality, a Kruskal-Wallis test was employed, followed by a Bonferroni-adjusted Dunn’s post-hoc test. A P-value of ≤ 0.05 was considered significant.
Results
Concordance between mini-exon sequence diversity and DTU identity
The monoclonal reference strains included in the study were Chilec22 cl.6 (TcI), M5521 cl.3 and cl.5 (TcIII), Arma18 cl.1 and cl.5 (TcIII), Saimiri3 cl.8 (TcIV), Para6 cl.1 (TcV), Para7 cl.3 (TcVI), Chaco9 cl.15 (TcVI) and LHVA cl.1 (TcVI) [10, 50, 64]. The mini-exon gene sequence was analyzed for each of these monoclonal reference strains, revealing a predominant haplotype (~98% of sequences) with one or few different sequences in most samples (Fig. 2).

Distinctive sequence types derived from monoclonal samples. Monoclonal samples from reference strains are displayed on the x-axis. The y-axis includes distinctive sequence types identified in this study that showed association with reference strains (haplotypes seen almost exclusively within a single monoclonal sample as representative of a DTU). Color intensity is proportional to the number of reads, here standardized for each row (sequence/haplotype) and displayed as a z-score
Genotyping by multiplex PCR and NGS
Among the 48 samples analyzed by multiplex PCR, agarose electrophoresis patterns indicated that 37.5% (n = 18) of the samples were co-infected with T. cruzi and T. rangeli, while 52.1% (n = 25) presented infection exclusively with T. cruzi and 6.25% (n = 3) were infected only with T. rangeli. Samples TBM2873 and TBM2983 showed faint bands in agarose gels and no clear pattern was obtained. Meanwhile, NGS was performed only on 43 samples from the Ecuadorian dataset; 5 were not sequenced due to technical issues. An additional 9 samples were excluded from further analysis because they did not reach the 20,000 reads threshold, indicating insufficient sequencing depth. Among the 34 samples for which NGS was successfully performed, taxonomic assignment in GenBank identified sequence types associated with both T. cruzi and T. rangeli in 25, while 8 were deemed to contain exclusively T. cruzi-like sequences, and only one had exclusively T. rangeli-like sequences (TBR1455). Remarkably, sequence types matching DTU TcIV were found in samples TBR1410, TBR1422, TBR1445, TBR1475, TBR1492, TBR1510, TBM2873, TBM2903 and TBM2983 (Table 1, Fig. 3).

Sequence types associated with T. cruzi and/or T. rangeli amplified from intestinal content of Ecuadorian triatomines. The y-axis shows the sequence types and their assigned DTU according to their similarity with GenBank reference sequences (> 98%). Samples are shown in the x-axis, indicating their assigned species and DTU. Color intensity is proportional to the number of reads, here standardized for each row (sequence/haplotype) and displayed as a z-score. * Indicates that the number of reads for these samples is too low to be visualized in the z-score adjusted heatmap. Among these, ** TBM2873 is the only sample where a TcI/TcIV co-infection was detected
Mini-exon sequence types distinctively associated with monoclonal strains TcI, TcIII, TcIV and TcV/VI were used for the construction of a neighbor-joining (NJ) tree alongside selected sequence types derived from Ecuadorian strains and T. rangeli. Haplotypes with more than 10,000 reads were selected, although key sequences associated with a specific DTU and Ecuadorian TcIV representatives were included in the alignment (Additional file 3: Alignment S1). The tree topology shows five robustly supported clusters, one corresponding to T. rangeli, separated from four corresponding to T. cruzi. The TcI-like sequences cluster together in one branch, separated from TcIV-like, TcIII-like and TcII/V/VI-like sequences (Fig. 4).

NJ analysis of selected mini-exon sequence types derived from Ecuadorian samples and monoclonal reference strains. Rooted tree (user selection) generated in MEGA7. Alignment performed by Muscle and manually edited. All haplotypes showing > 10,000 reads were included. Other haplotypes with < 10,000 reads were included to ensure all DTUs were represented
NJ analysis including Ecuadorian T. rangeli haplotypes yielded a rather uninformative clustering with several polytomies. Therefore, only selected T. rangeli haplotypes were included along with available sequence data retrieved from NCBI (Fig. 5, Additional file 4: Alignment S2, Additional file 5: Table S3). The Ecuadorian haplotypes clustered with KP1(-)/C reference sequences. Although bootstrap values are low for most branches, KP1(+), KP1(−) and groups A, B, C and E [34] are also evident in the tree.

NJ analysis of selected T. rangeli haplotypes derived from Ecuadorian samples. Rooted tree (user selection) generated in MEGA7. Alignment performed by Muscle and manually edited. Reference sequences available at the NCBI database are included (Additional file 4: Alignment S2 shows the alignment used for tree generation and Additional file 5: Table S3 shows the sequences retrieved from the NCBI database)
Parasite richness and diversity
Chao richness and Shannonʼs diversity indices were calculated for each parasite species within each sample. No significant differences in richness or diversity were found between samples from the two studied localities (Bramaderos and Bellamaria) for either parasite species. Analyses were performed merging both male and female adults and with adults from each sex taken independently. In both cases, we found significant differences for T. rangeli richness across developmental stages (Kruskal-Wallis H-test: χ2 = 8.4478, df = 3, P = 0.0376 for male/female independent analysis, and Kruskal-Wallis H-test: χ2 = 6.8417, df = 2, P = 0.0327 for the merged group). The Bonferroni-adjusted Dunn’s post-hoc test showed significant differences between instar V nymphs and adult females (P = 0.0124) and among instar V nymphs and the merged group of adult females and males (P = 0.0138). In both cases, instar V showed significantly higher richness than the remaining developmental stages.
Discussion
Comparison of parasite identification/genotyping methods
We compared the results from three different types of molecular analysis for 48 intestinal content DNA samples from triatomine bugs collected in two rural communities of Loja Province, southern Ecuador. All samples selected for the study had been previously characterized as infected with T. cruzi based on PCR product size polymorphism of kinetoplast minicircle sequences, with no evidence of co-infection with the sympatric sister species T. rangeli. The sample set was subsequently analyzed by two additional molecular techniques; first by PCR product size polymorphism analysis of the non-transcribed spacer region of the mini-exon gene, then by deep sequencing of these mini-exon amplicons. Incongruences were detected among the results obtained by the three methods, and our results suggest that simple amplicon visualization of kinetoplast minicircle sequences may not suffice for detection of T. cruzi + T. rangeli co-infections.
The PCR-based method for mini-exon gene analysis without sequencing [47] uncovered 18 instances of T. cruzi + T. rangeli co-infections in our sample panel, which the kinetoplast minicircle PCR [58] did not identify. Mixed infections of T. cruzi and T. rangeli in vector and mammalian hosts are frequent [18, 23, 65, 66] and several groups have reported that minicircle kinetoplast PCR product visualization techniques are inadequate to identify them [66,67,68,69]. Previous studies have applied this same method in southern Ecuador and reported ~10% prevalence of T. rangeli infection in sampled triatomines and mammals and just ~1.25% in co-infection with T. cruzi [29]. In stark contrast, NGS revealed T. rangeli-like sequences in 26 out of 34 (76.47%) infected samples characterized in our study.
Trypanosoma rangeli and T. cruzi genetic diversity
Our sample panel was heavily biased toward R. ecuadoriensis (45 out of 48 DNA samples were isolated from this vector species), one of the two most important vectors of CD in the study region and in Ecuador at large [70]. A close association between T. rangeli and Rhodnius spp. has been established in the past, where 12 out of 15 species of the genus have shown vectorial capacity [26]. Our results suggest that previous reports regarding T. rangeli infection in R. ecuadoriensis may underestimate the presence of this parasite in southern Ecuador, where a wider distribution of T. rangeli in Loja, and perhaps the rest of the country, may have been overlooked. Furthermore, although only a ~100-bp fragment was analyzed, a wide T. rangeli genetic diversity was encountered (79 haplotypes in 26 triatomines). Consistent with previous reports, where 99% of Ecuadorian T. rangeli were classified as KP1(−), and 80% as lineage C [29], NJ analysis clustered all Ecuadorian sample haplotypes with KP1(-)/C reference sequences. Low bootstrap values in this analysis may arise from the small size (~100 bp) of the T. rangeli amplicon and the limited number of informative SNPs found within the sequence (some haplotypes differ by only one SNP). Notably, we did not identify any KP1(+) haplotypes, although previous reports indicate KP1(+) and (−) lineages are sympatric and may be found in the intestinal contents of Rhodnius spp. [71].
Co-infection with T. cruzi and T. rangeli occurred in 73.5% of analyzed triatomines. Despite earlier reports suggesting T. rangeli infection is pathogenic to triatomines, more recent studies point to the need of more solid evidence before generalizing this claim [72]. Indeed, recent reports suggest co-infection increases R. prolixus survival, reproduction and fitness, which in turn would favor transmission of both parasite species [73]. The interplay of the infection with these two trypanosomatids in R. ecuadoriensis and its possible impact over the epidemiology of Chagas disease in the south of Ecuador warrants further investigation.
Sequencing of the non-transcribed spacer of the mini-exon gene has proven useful in phylogenetic analysis and has provided valuable insights for DTU discrimination while analyzing T. cruzi intra-strain variability [10, 48, 51, 55]. Recently, deep sequencing of the non-transcribed spacer has revealed high levels of T. cruzi diversity in the intestinal content of T. dimidiata [57]. Similarly, Herrera et al. [49] recently employed NGS sequencing of the mini-exon gene to analyze the genetic diversity of T. cruzi naturally infecting a group of captive non-human primates in the southern USA. In our study, 14.5 T. cruzi and 14.4 T. rangeli haplotypes were detected on average per triatomine. However, we do note from our analysis of monoclonal reference samples that mini-exon sequences may not accurately resolve T. cruzi phylogenetic diversity, thus reflecting to an extent the lack of reliability of this locus in identifying DTUs. From monoclonal strain haplotypes, our data showed that some low abundance sequence types are shared by almost all DTUs, with important implications for those using the mini-exon splice leader locus to define intra-host DTU diversity (Additional file 1: Table S1). Therefore, it is also difficult to say with confidence that the high levels of T. cruzi diversity found in our samples result from multiclonal infections, given that multiple divergent copies are present per genome. Nonetheless, a subset of high abundance sequence types could be associated with different DTUs and it is upon these that we base our discrimination between TcI and TcIV in this study.
TcI is the predominant DTU in Ecuador [13,14,15, 23]. As expected, TcI-like haplotypes were widely represented in the deep sequencing analysis of the mini-exon gene performed for the Ecuadorian samples. Notably, we identified sequences matching DTU TcIV in nine of the Ecuadorian samples. Two TcIV-like haplotypes displayed a 99% match with an NCBI entry originated from a TcIV isolate. The other two TcIV-like haplotypes found also showed high homology (97% and 98%). Additionally, NJ analysis clustered the Ecuadorian TcIV haplotypes with discriminatory TcIV sequences available from monoclonal strains with known DTU identity. Strong bootstrap values support this finding; therefore, we are confident of the identification of TcIV haplotypes in our sample panel. To the best of our knowledge, this constitutes the first report of TcIV in Loja Province and is a testament to the power of amplicon sequencing to uncover ‘hidden’ T. cruzi diversity. Only two previous reports of DTUs different from TcI in Ecuador exist. One of them, from the early 2000s, employed multilocus enzyme electrophoresis to identify zymodemes II and III [16]. The second report provided serological evidence using a TcII/V/VI-specific epitope [17]. Our findings expand the distribution of DTU TcIV to the southernmost province of Ecuador, where its presence had not previously been reported.
No major differences were found between the studied localities in terms of parasite genetic richness or diversity. Both communities are only 28.3 km apart and infected livestock, small mammals, passive transportation of triatomines or human movement may cause active parasite dispersal, homogenizing the parasite populations. In addition, no significant differences in T. cruzi richness or diversity were detected across vector developmental stages. The hostile environment in the vector’s anterior midgut has been previously suggested to create a bottleneck [74], which could reduce the diversity of T. cruzi. In contrast, the significantly higher T. rangeli richness found in instar V compared to adult triatomines is consistent with the notion of this parasite being pathogenic for triatomines [75, 76], which could affect their development into adulthood.
Dumonteil et al. [57] reported evidence of T. cruzi clone accumulation in T. dimidiata as a result of subsequent feeding events by this vector. The data do not support the occurrence of such accumulation in our sample set, which may be linked to the presence of multiple copies, and variants, of mini-exon sequences within clones, which obscure the signal of accumulating clonal diversity over multiple infected blood meals.
Conclusions
Our results indicate that techniques based solely on PCR amplification of minicircle or mini-exon sequences are not optimal for detection and characterization of mixed infections with kinetoplastid parasites in triatomines. Employing deep sequencing of the non-transcribed spacer region of the mini-exon gene, we have encountered 73.5% (considering the 34 samples with NGS results) of studied triatomines to be co-infected with T. cruzi and T. rangeli, a much higher prevalence for T. rangeli infection than previously reported for vectors in the study region. Haplotypes of T. rangeli found in Ecuadorian samples were characterized as KP1(-)/C, in congruence with previous reports. Additionally, we provide the first report for the presence of DTU TcIV in Loja Province. In conclusion, we demonstrate the power of NGS to scrutinize parasite populations with high resolution. However, we do note that the mini-exon marker must be approached with caution, and further deep sequencing analyses targeted at additional markers, potentially conserved house-keeping genes [77], are required for confirmation. Nonetheless, the information presented in this report is of value in the characterization of the dynamics of T. cruzi transmission in southern Ecuador and must be considered in disease control efforts in the region.
Availability of data and materials
Data supporting the conclusions of this article are included within the article and its additional files. The datasets generated during this study are available at the sequence read archive (SRA) database, bioproject PRJNA596271, https://www.ncbi.nlm.nih.gov/sra/PRJNA596271. The alignment used for Fig. 4 is included in Additional file 3: Alignment S1, and the alignment used for Fig. 5 is included in Additional file 4: Alignment S2.
Abbreviations
- CD:
-
Chagas disease
- DTU:
-
discrete typing unit
- TcI:
-
Trypanosoma cruzi I
- PCR:
-
polymerase chain reaction
- RAPD:
-
random amplification of polymorphic DNA
- SSU rRNA:
-
small subunit of ribosomal RNA
- ITS rDNA:
-
internal transcribed spacer of rDNA
- SNP:
-
single-nucleotide polymorphism
- NGS:
-
next-generation sequencing
- CISeAL:
-
Spanish acronym for the Center for Research on Health in Latin America
- IGM:
-
Spanish acronym for the Ecuadorian Military Geographic Institute
- GPS:
-
global positioning service
- BLAST:
-
Basic Local Alignment Search Tool
- NCBI:
-
National Center for Biotechnology Information
- ANOVA:
-
analysis of variance
- NJ:
-
neighbor-joining
- SRA:
-
sequence read archive database
References
Martinez F, Perna E, Perrone SV, Liprandi AS. Chagas disease and heart failure: an expanding issue worldwide. Eur Cardiol. 2019;14:82–8.
Lindani KCF, Andrade FA, Bavia L, Damasceno FS, Beltrame MH, Messias-Reason IJ, Sandri TL. Chagas disease: from discovery to a worldwide health problem. Front Public Health. 2019;7:166.
Coura JR, Viñas PA. Chagas disease: a new worldwide challenge. Nature. 2010;465:S6–7.
Miles MA, Souza A, Povoa M, Shaw JJ, Lainson R, Toye PJ. Isozymic heterogeneity of Trypanosoma cruzi in the first autochthonous patients with Chagas’ disease in Amazonian Brazil. Nature. 1978;272:819–21.
Miles MA, Cedillos RA, Póvoa MM, de Souza AA, Prata A, Macedo V. Do radically dissimilar Trypanosoma cruzi strains (zymodemes) cause Venezuelan and Brazilian forms of Chagas’ disease? Lancet. 1981;1:1338–40.
Tibayrenc M, Ward P, Moya A, Ayala FJ. Natural populations of Trypanosoma cruzi, the agent of Chagas disease, have a complex multiclonal structure. Proc Natl Acad Sci USA. 1986;83:115–9.
Tibayrenc M. Genetic epidemiology of parasitic protozoa and other infectious agents: the need for an integrated approach. Int J Parasitol. 1998;28:85–104.
Brisse S, Dujardin JC, Tibayrenc M. Identification of six Trypanosoma cruzi lineages by sequence-characterized amplified region markers. Mol Biochem Parasitol. 2000;111:95–105.
Brisse S, Barnabé C, Tibayrenc M. Identification of six Trypanosoma cruzi phylogenetic lineages by random amplified polymorphic DNA and multilocus enzyme electrophoresis. Int J Parasitol. 2000;30:35–44.
Zingales B, Andrade SG, Briones MRS, Campbell DA, Chiari E, Fernandes O, et al. A new consensus for Trypanosoma cruzi intraspecific nomenclature: second revision meeting recommends TcI to TcVI. Mem Inst Oswaldo Cruz. 2009;104:1051–4.
Marcili A, Lima L, Cavazzana M, Junqueira ACV, Veludo HH, Silva FMD, et al. A new genotype of Trypanosoma cruzi associated with bats evidenced by phylogenetic analyses using SSU rDNA, cytochrome b and histone H2B genes and genotyping based on ITS1 rDNA. Parasitology. 2009;136:641–55.
Lima L, Espinosa-Álvarez O, Ortiz PA, Trejo-Varón JA, Carranza JC, Pinto CM, et al. Genetic diversity of Trypanosoma cruzi in bats, and multilocus phylogenetic and phylogeographical analyses supporting Tcbat as an independent DTU (discrete typing unit). Acta Trop. 2015;151:166–77.
Wong YY, Sornosa Macias KJ, Guale Martínez D, Solorzano LF, Ramirez-Sierra MJ, Herrera C, et al. Molecular epidemiology of Trypanosoma cruzi and Triatoma dimidiata in costal Ecuador. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis. 2016;41:207–12.
Costales JA, Jara-Palacios MA, Llewellyn MS, Messenger LA, Ocaña-Mayorga S, Villacís AG, et al. Trypanosoma cruzi population dynamics in the Central Ecuadorian Coast. Acta Trop. 2015;151:88–93.
Ocaña-Mayorga S, Llewellyn MS, Costales JA, Miles MA, Grijalva MJ. Sex, subdivision, and domestic dispersal of Trypanosoma cruzi lineage I in southern Ecuador. PLoS Negl Trop Dis. 2010;4:e915.
Garzón EA, Barnabé C, Córdova X, Bowen C, Paredes W, Gómez E, et al. Trypanosoma cruzi isoenzyme variability in Ecuador: first observation of zymodeme III genotypes in chronic chagasic patients. Trans R Soc Trop Med Hyg. 2002;96:378–82.
Bhattacharyya T, Falconar AK, Luquetti AO, Costales JA, Grijalva MJ, Lewis MD, et al. Development of peptide-based lineage-specific serology for chronic Chagas disease: geographical and clinical distribution of epitope recognition. PLoS Negl Trop Dis. 2014;8:e2892.
Villacís AG, Ocaña-Mayorga S, Lascano MS, Yumiseva CA, Baus EG, Grijalva MJ. Abundance, natural infection with trypanosomes, and food source of an endemic species of triatomine, Panstrongylus howardi (Neiva 1911), on the Ecuadorian Central Coast. Am J Trop Med Hyg. 2015;92:187–92.
Grijalva MJ, Villacis AG, Ocaña-Mayorga S, Yumiseva CA, Moncayo AL, Baus EG. Comprehensive survey of domiciliary triatomine species capable of transmitting Chagas disease in southern Ecuador. PLoS Negl Trop Dis. 2015;9:e0004142.
Díaz S, Panzera F, Jaramillo-O N, Pérez R, Fernández R, Vallejo G, et al. Genetic, cytogenetic and morphological trends in the evolution of the Rhodnius (Triatominae: Rhodniini) trans-Andean group. PLoS One. 2014;9:e87493.
Abad-Franch F, Aguilar VHM, Paucar CA, Lorosa ES, Noireau F. Observations on the domestic ecology of Rhodnius ecuadoriensis (Triatominae). Mem Inst Oswaldo Cruz. 2002;97:199–202.
Guhl F, Hudson L, Marinkelle CJ, Jaramillo CA, Bridge D. Clinical Trypanosoma rangeli infection as a complication of Chagas’ disease. Parasitology. 1987;94:475–84.
Grijalva MJ, Suarez-Davalos V, Villacis AG, Ocaña-Mayorga S, Dangles O. Ecological factors related to the widespread distribution of sylvatic Rhodnius ecuadoriensis populations in southern Ecuador. Parasit Vectors. 2012;5:17.
Guhl F, Hudson L, Marinkelle CJ, Morgan SJ, Jaramillo C. Antibody response to experimental Trypanosoma rangeli infection and its implications for immunodiagnosis of South American trypanosomiasis. Acta Trop. 1985;42:311–8.
Zuniga C, Vargas R, Palau MT, Bello F, Diego D, Antonio J, et al. Trypanosoma rangeli infected mouse sera reactivity with Trypanosoma cruzi synthetic peptides. Parasitol Latinoam. 2007;62:3–6.
Guhl F, Vallejo GA. Trypanosoma (Herpetosoma) rangeli Tejera, 1920: an updated review. Mem Inst Oswaldo Cruz. 2003;98:435–42.
Silva FMD, Rodrigues AC, Campaner M, Takata CSA, Brigido MC, Junqueira ACV, et al. Randomly amplified polymorphic DNA analysis of Trypanosoma rangeli and allied species from human, monkeys and other sylvatic mammals of the Brazilian Amazon disclosed a new group and a species-specific marker. Parasitology. 2004;128:283–94.
Garcia ES, Castro DP, Figueiredo MB, Azambuja P. Parasite-mediated interactions within the insect vector: Trypanosoma rangeli strategies. Parasit Vectors. 2012;5:105.
Ocaña-Mayorga S, Aguirre-Villacis F, Pinto CM, Vallejo GA, Grijalva MJ. Prevalence, genetic characterization, and 18S small subunit ribosomal RNA diversity of Trypanosoma rangeli in triatomine and mammal hosts in endemic areas for Chagas disease in Ecuador. Vector Borne Zoonotic Dis. 2015;15:732–42.
Degrave W, Fragoso SP, Britto C, van Heuverswyn H, Kidane GZ, Cardoso MA, et al. Peculiar sequence organization of kinetoplast DNA minicircles from Trypanosoma cruzi. Mol Biochem Parasitol. 1988;27:63–70.
Recinos RF, Kirchhoff LV, Donelson JE. Characterization of kinetoplast DNA minicircles in Trypanosoma rangeli. Mol Biochem Parasitol. 1994;63:59–67.
Vallejo GA, Macedo AM, Chiari E, Pena SD. Kinetoplast DNA from Trypanosoma rangeli contains two distinct classes of minicircles with different size and molecular organization. Mol Biochem Parasitol. 1994;67:245–53.
Maia Da Silva F, Junqueira ACV, Campaner M, Rodrigues AC, Crisante G, Ramirez LE, et al. Comparative phylogeography of Trypanosoma rangeli and Rhodnius (Hemiptera: Reduviidae) supports a long coexistence of parasite lineages and their sympatric vectors. Mol Ecol. 2007;16:3361–73.
Maia da Silva F, Marcili A, Lima L, Cavazzana M, Ortiz PA, Campaner M, et al. Trypanosoma rangeli isolates of bats from Central Brazil: genotyping and phylogenetic analysis enable description of a new lineage using spliced-leader gene sequences. Acta Trop. 2009;109:199–207.
Sincero TCM, Stoco PH, Steindel M, Vallejo GA, Grisard EC. Trypanosoma rangeli displays a clonal population structure, revealing a subdivision of KP1(-) strains and the ancestry of the Amazonian group. Int J Parasitol. 2015;45:225–35.
Bosseno MF, Telleria J, Vargas F, Yaksic N, Noireau F, Morin A, et al. Trypanosoma cruzi: study of the distribution of two widespread clonal genotypes in Bolivian Triatoma infestans vectors shows a high frequency of mixed infections. Exp Parasitol. 1996;83:275–82.
Solari A, Campillay R, Ortíz S, Wallace A. Identification of Trypanosoma cruzi genotypes circulating in Chilean chagasic patients. Exp Parasitol. 2001;97:226–33.
Yeo M, Lewis MD, Carrasco HJ, Acosta N, Llewellyn M, da Silva Valente SA, et al. Resolution of multiclonal infections of Trypanosoma cruzi from naturally infected triatomine bugs and from experimentally infected mice by direct plating on a sensitive solid medium. Int J Parasitol. 2007;37:111–20.
Llewellyn MS, Rivett-Carnac JB, Fitzpatrick S, Lewis MD, Yeo M, Gaunt MW, et al. Extraordinary Trypanosoma cruzi diversity within single mammalian reservoir hosts implies a mechanism of diversifying selection. Int J Parasitol. 2011;41:609–14.
Perez CJ, Lymbery AJ, Thompson RCA. Chagas disease: the challenge of polyparasitism? Trends Parasitol. 2014;30:176–82.
Ramírez JD, Guhl F, Messenger LA, Lewis MD, Montilla M, Cucunuba Z, et al. Contemporary cryptic sexuality in Trypanosoma cruzi. Mol Ecol. 2012;21:4216–26.
Taylor SM, Parobek CM, Aragam N, Ngasala BE, Mårtensson A, Meshnick SR, et al. Pooled deep sequencing of Plasmodium falciparum isolates: an efficient and scalable tool to quantify prevailing malaria drug-resistance genotypes. J Infect Dis. 2013;208:1998–2006.
Bernards A, Van der Ploeg LH, Frasch AC, Borst P, Boothroyd JC, Coleman S, et al. Activation of trypanosome surface glycoprotein genes involves a duplication-transposition leading to an altered 3’ end. Cell. 1981;27:497–505.
De Lange T, Liu AY, Van der Ploeg LH, Borst P, Tromp MC, Van Boom JH. Tandem repetition of the 5’ mini-exon of variant surface glycoprotein genes: a multiple promoter for VSG gene transcription? Cell. 1983;34:891–900.
Borst P. Discontinuous transcription and antigenic variation in trypanosomes. Annu Rev Biochem. 1986;55:701–32.
Murthy VK, Dibbern KM, Campbell DA. PCR amplification of mini-exon genes differentiates Trypanosoma cruzi from Trypanosoma rangeli. Mol Cell Probes. 1992;6:237–43.
Fernandes O, Santos SS, Cupolillo E, Mendonça B, Derre R, Junqueira ACV, et al. A mini-exon multiplex polymerase chain reaction to distinguish the major groups of Trypanosoma cruzi and T. rangeli in the Brazilian Amazon. Trans R Soc Trop Med Hyg. 2001;95:97–9.
O’Connor O, Bosseno M-F, Barnabé C, Douzery EJP, Brenière SF. Genetic clustering of Trypanosoma cruzi I lineage evidenced by intergenic miniexon gene sequencing. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis. 2007;7:587–93.
Herrera C, Majeau A, Didier P, Falkenstein KP, Dumonteil E. Trypanosoma cruzi diversity in naturally infected nonhuman primates in Louisiana assessed by deep sequencing of the mini-exon gene. Trans R Soc Trop Med Hyg. 2019;113:281–6.
Lewis MD, Ma J, Yeo M, Carrasco HJ, Llewellyn MS, Miles MA. Genotyping of Trypanosoma cruzi: systematic selection of assays allowing rapid and accurate discrimination of all known lineages. Am J Trop Med Hyg. 2009;81:1041–9.
Herrera C, Bargues MD, Fajardo A, Montilla M, Triana O, Vallejo GA, et al. Identifying four Trypanosoma cruzi I isolate haplotypes from different geographic regions in Colombia. Infect Genet Evol. 2007;7:535–9.
Herrera C, Guhl F, Falla A, Fajardo A, Montilla M, Adolfo Vallejo G, et al. Genetic variability and phylogenetic relationships within Trypanosoma cruzi I isolated in Colombia based on miniexon gene sequences. J Parasitol Res. 2009;2009:897364.
Falla A, Herrera C, Fajardo A, Montilla M, Vallejo GA, Guhl F. Haplotype identification within Trypanosoma cruzi I in Colombian isolates from several reservoirs, vectors and humans. Acta Trop. 2009;110:15–21.
Herrera CP, Barnabé C, Brenière SF. Complex evolutionary pathways of the intergenic region of the mini-exon gene in Trypanosoma cruzi TcI: a possible ancient origin in the Gran Chaco and lack of strict genetic structuration. Infect Genet Evol. 2013;16:27–37.
Martínez I, Nogueda B, Martinez-Hernandez F, Espinoza B. Microsatellite and mini-exon analysis of Mexican human DTU I Trypanosoma cruzi strains and their susceptibility to nifurtimox and benznidazole. Vector Borne Zoonotic Dis. 2013;13:181–7.
Llewellyn MS, Messenger LA, Luquetti AO, Garcia L, Torrico F, Tavares SBN, et al. Deep sequencing of the Trypanosoma cruzi GP63 surface proteases reveals diversity and diversifying selection among chronic and congenital Chagas disease patients. PLoS Negl Trop Dis. 2015;9:e0003458.
Dumonteil E, Ramirez-Sierra MJ, Pérez-Carrillo S, Teh-Poot C, Herrera C, Gourbière S, et al. Detailed ecological associations of triatomines revealed by metabarcoding and next-generation sequencing: implications for triatomine behavior and Trypanosoma cruzi transmission cycles. Sci Rep. 2018;8:4140.
Wincker P, Britto C, Pereira JB, Cardoso MA, Oelemann W, Morel CM. Use of a simplified polymerase chain reaction procedure to detect Trypanosoma cruzi in blood samples from chronic chagasic patients in a rural endemic area. Am J Trop Med Hyg. 1994;51:771–7.
Ison SA, Delannoy S, Bugarel M, Nagaraja TG, Renter DG, den Bakker HC, et al. Targeted amplicon sequencing for single-nucleotide-polymorphism genotyping of attaching and effacing Escherichia coli O26:H11 cattle strains via a high-throughput library preparation technique. Appl Environ Microbiol. 2016;82:640–9.
Joshi NA, Fass JN. Sickle: Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33); 2011. https://github.com/najoshi/sickle. Accessed 15 Feb 2018.
Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for Illumina sequences. BMC Bioinformatics. 2012;13:31.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1.
Shannon CE, Weaver W. The mathematical theory of communication. Champaign: University of Illinois Press; 1971.
Yeo M, Acosta N, Llewellyn M, Sánchez H, Adamson S, Miles GAJ, et al. Origins of Chagas disease: Didelphis species are natural hosts of Trypanosoma cruzi I and armadillos hosts of Trypanosoma cruzi II, including hybrids. Int J Parasitol. 2005;35:225–33.
Grijalva M, Villacis A. Presence of Rhodnius ecuadoriensis in sylvatic habitats in the southern highlands (Loja Province) of Ecuador. J Med Entomol. 2009;46:708–11.
Vargas N, Souto RP, Carranza JC, Vallejo GA, Zingales B. Amplification of a specific repetitive DNA sequence for Trypanosoma rangeli identification and its potential application in epidemiological investigations. Exp Parasitol. 2000;96:147–59.
Vallejo GA, Guhl F, Chiari E, Macedo AM. Species specific detection of Trypanosoma cruzi and Trypanosoma rangeli in vector and mammalian hosts by polymerase chain reaction amplification of kinetoplast minicircle DNA. Acta Trop. 1999;72:203–12.
Ramirez LE, Lages-Silva E, Alvarenga-Franco F, Matos A, Vargas N, Fernandes O, et al. High prevalence of Trypanosoma rangeli and Trypanosoma cruzi in opossums and triatomids in a formerly-endemic area of Chagas disease in Southeast Brazil. Acta Trop. 2002;84:189–98.
Pavia PX, Vallejo GA, Montilla M, Nicholls RS, Puerta CJ. Detection of Trypanosoma cruzi and Trypanosoma rangeli infection in triatomine vectors by amplification of the histone H2A/SIRE and the sno-RNA-C11 genes. Rev Inst Med Trop São Paulo. 2007;49:23–30.
Villacís AG, Marcet PL, Yumiseva CA, Dotson EM, Tibayrenc M, Brenière SF, et al. Pioneer study of population genetics of Rhodnius ecuadoriensis (Hemiptera: Reduviidae) from the central coastand southern Andean regions of Ecuador. Infect Genet Evol. 2017;53:116–27.
Vallejo GA, Guhl F, Carranza JC, Lozano LE, Sánchez JL, Jaramillo JC, et al. kDNA markers define two major Trypanosoma rangeli lineages in Latin-America. Acta Trop. 2002;81:77–82.
Peterson JK, Graham AL. What is the ‘true’ effect of Trypanosoma rangeli on its traitomine bug vector? J Vector Ecol. 2016;41:27–33.
Peterson JK, Graham AL, Elliott RJ, Dobson AP, Chávez OT. Trypanosoma cruzi-Trypanosoma rangeli co-infection ameliorates negative effects of single trypanosome infections in experimentally infected Rhodnius prolixus. Parasitology. 2016;143:1157–67.
Ferreira RC, Kessler RL, Lorenzo MG, Paim RMM, Ferreira LDL, Probst CM, et al. Colonization of Rhodnius prolixus gut by Trypanosoma cruzi involves an extensive parasite killing. Parasitology. 2016;143:434–43.
Añez N, Añez N. Studies on Trypanosoma rangeli Tejera 1920. VII—its effect on the survival of infected triatomine bugs. Mem Inst Oswaldo Cruz. 1920;1984(79):249–55.
Fellet MR, Lorenzo MG, Elliot SL, Carrasco D, Guarneri AA. Effects of infection by Trypanosoma cruzi and Trypanosoma rangeli on the reproductive performance of the vector Rhodnius prolixus. PLoS One. 2014;9:e105255.
Yeo M, Mauricio IL, Messenger LA, Lewis MD, Llewellyn MS, Acosta N, et al. Multilocus sequence typing (MLST) for lineage assignment and high resolution diversity studies in Trypanosoma cruzi. PLoS Negl Trop Dis. 2011;5:e1049.
Acknowledgements
Special thanks to Anita Villacís from CISeAL’s Medical Entomology Unit; to Alejandra Zurita and Sofía Ocaña for parasite and DNA isolation, information regarding infection status based on kinetoplast PCR gel electrophoresis, and associated metadata; and to César Yumiseva for assistance in the generation of maps. Special thanks to Julie Galbraith for assistance with next-generation sequencing, and to Camila Cilveti, Christopher Garcia and Andrea Vela from CISeAL.
Funding
This study was funded by the National Institutes of Health (http://www.nih.gov), grant number: R15 AI105749-01A1, awarded to MJG. This work was carried out as part of JMS’s Master’s degree, whose stipend was funded by the Fogarty International Center-National Institutes of Health (https://www.fic.nih.gov), through the Global Infectious Diseases Training Program, grant number: D43TW008261, awarded to MJG and JAC. Samples employed in the study were collected in previous projects, funded by Pontificia Universidad Católica del Ecuador (projects I13048, J13049). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
MSL, MJG and JAC designed the study. JMS and PS performed experimental procedures. JMS, PS and SOBS analyzed the data. JMS, PS and JAC interpreted the results. JMS, JAC prepared the manuscript. All authors edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Raw data for monoclones and Ecuadorian samples. Haplotype ID, identity and number of reads are included.
Additional file 2: Table S2.
Correspondence between FLDs* (Illumina sequencer output) and samples belonging to intestinal contents.
Additional file 3: Alignment S1.
Sequences employed for the phylogenetic tree displayed in Fig. 4.
Additional file 4: Alignment S2.
Sequences employed for the phylogenetic tree displayed in Fig. 5.
Additional file 5: Table S3.
Accession numbers of downloaded sequences from NCBI database used in the generation of the NJ tree for T. rangeli. The retrieved sequences were trimmed to match with the amplicons generated with Ecuadorian samples. The discarded portions of the sequences are shown. Lineage (A, B, C, D and E) and KP1(+/−) information was included when available. Geographical origin of the samples is also in the file.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Maiguashca Sánchez, J., Sueto, S.O.B., Schwabl, P. et al. Remarkable genetic diversity of Trypanosoma cruzi and Trypanosoma rangeli in two localities of southern Ecuador identified via deep sequencing of mini-exon gene amplicons. Parasites Vectors 13, 252 (2020). https://doi.org/10.1186/s13071-020-04079-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-020-04079-1