
Abstract
Background
Biting midge species of the genus Culicoides Latreille (Diptera: Ceratopogonidae) comprise more than 1300 species distributed worldwide. Several species of Culicoides are vectors of various viruses that can affect animals, like the African horse sickness virus (AHSV), known to be endemic in sub-Saharan Africa. The ecological and veterinary interest of Culicoides emphasizes the need for rapid and reliable identification of vector species. However, morphology-based identification has limitations and warrants integration of molecular data. DNA barcoding based on the mitochondrial gene cytochrome c oxidase subunit 1 (cox1) is used as a rapid and authentic tool for species identification in a wide variety of animal taxa across the globe. In this study, our objectives were as follows: (i) establish a reference DNA barcode for Afrotropical Culicoides species; (ii) assess the accuracy of cox1 in identifying Afrotropical Culicoides species; and (iii) test the applicability of DNA barcoding for species identification on a large number of samples of Culicoides larvae from the Niayes area of Senegal, West Africa.
Results
A database of 230 cox1 sequences belonging to 42 Afrotropical Culicoides species was found to be reliable for species-level assignments, which enabled us to identify cox1 sequences of Culicoides larvae from the Niayes area of Senegal. Of the 933 cox1 sequences of Culicoides larvae analyzed, 906 were correctly identified by their barcode sequences corresponding to eight species of Culicoides. A total of 1131 cox1 sequences of adult and larval Culicoides were analyzed, and a hierarchical increase in mean divergence was observed according to two taxonomic levels: within species (mean = 1.92%, SE = 0.00), and within genus (mean = 17.82%, SE = 0.00).
Conclusions
Our study proves the efficiency of DNA barcoding for studying Culicoides larval diversity in field samples. Such a diagnostic tool offers great opportunities for investigating Culicoides immature stages ecology and biology, a prerequisite for the implementation of eco-epidemiological studies to better control AHSV in the Niayes region of Senegal, and more generally in sub-Saharan Africa.
Similar content being viewed by others


Background
Biting midge species of the genus Culicoides Latreille (Diptera: Ceratopogonidae) comprise more than 1300 species distributed worldwide [1]. Certain Culicoides species are the biological vectors of important arboviruses of livestock worldwide, such as the African horse sickness virus (AHSV), bluetongue virus (BTV), epizootic hemorrhagic disease virus (EHDV), equine encephalosis virus (EEV) and Schmallenberg virus (SBV) [2]. African horse sickness virus is an arbovirus of equids that is biologically transmitted by competent vectors of the genus Culicoides [3]. This disease is recorded in Africa and Arabian Peninsula and is ranked among the most lethal of viral infections known to affect horses with mortality rates in naive equine populations that can reach 80–90% [3, 4]. Massive AHS epizootic outbreaks occurred in Senegal in 2007 [5, 6]. Knowledge on the ecology of Culicoides will be crucial for the development and implementation of appropriate and effective vector control strategies in order to reduce the impact of Culicoides-borne diseases. However, a major limitation is that morphology-based methods for Culicoides species identification are time-consuming and require taxonomic expertise. Adult morphological identification may involve dissection and microscopical mounting of specimens. Taking into account that subadult stages of the majority of Culicoides species still await discovery [7], morphological species identification of Culicoides larvae is not possible. Inaccurate Culicoides species identification can have significant impacts on control attempts.
Considering these difficulties, it is essential to use complementary and alternative methods to solve taxonomic problems such as the identification of Culicoides larvae. Although molecular tools may be expensive and require specialized equipment, they have been useful over the last decade to deepen knowledge in various areas of biology ranging from systematics to ecology [8,9,10,11,12,13]. Hebert et al. [9] proposed using the mitochondrial gene cytochrome c oxidase subunit 1 (cox1) as a DNA-based identification system for all animal species, the so-called DNA barcoding approach. DNA barcoding for species-level identification employs a small portion (≈ 658 bp) of the cox1 gene to assign a specimen sequence to a voucher species library [9]. This has gained wide acceptance as a supplementary method to resolve taxonomic ambiguities [9, 14]. However, successful DNA barcoding depends on the distinction between intra- and interspecific genetic divergence. The performance of DNA barcoding can vary within the same group of specimens among geographical regions and ecosystems [15]. Species with large effective population sizes can have high intraspecific genetic diversity, which could overlap with interspecific divergence [16]. Furthermore, imperfect taxonomy also could lead to erroneous identifications [17]. Therefore, morphological and molecular identification have both limitations and advantages, but in the absence of a large body of work on morphological identification of the Culicoides immature diversity in the Afrotropical region, advances in molecular identification would be a crucial stepping stone.
In the present study, our objectives were: (i) to establish DNA barcode libraries for adult Culicoides species collected in different sites in the Afrotropical region [18]; (ii) to assess the accuracy of the cox1 gene in identifying of these Culicoides species; and (iii) to test the usefulness of DNA barcoding for species identification on a large dataset of Culicoides larvae from the Niayes area of Senegal, West Africa. Our study establishes comprehensive DNA barcode libraries for Afrotropical Culicoides of interest prior to future taxonomic research such as metabarcoding.
Results
Reference DNA sequence analysis
Data description and distance summary
Haplotype data analysis detected 170 unique haplotypes in the DNA reference libraries (Table 1). The average nucleotide frequencies for all 42 species were as follows: A (adenine), 28%; T (thymine), 40%; G (guanine), 15.2%; and C (cytosine), 16.8%. The analysis revealed that interspecific Kimura-2-parameter (K2P) genetic divergence ranged between 0.045–0.201 with a mean genetic distance (MGD) of 0.133; intraspecific K2P genetic divergence ranged between 0–0.107 with an average of 0.009 (Table 1).
Identification success rates
In the simulations, the nearest-neighbour (NN) approach returned 97.39% correct and 2.61% incorrect identifications (Fig. 1). The threshold analysis (TA) returned the same results as best close match (BCM) at the threshold value 0.01 (79.56% correct and 20.44% incorrect identifications). With a threshold of 0.039 calculated by the function localMinima in SPIDER, the TA and BCM provided 94.68% correct and 5.32% incorrect identifications. With a threshold of 0.044 (Additional file 1: Figure S1) generated by the function threshVal in SPIDER, the TA and BCM provided 95.21% correct and 4.79% incorrect identifications. The proportion of monophyly on a neighbor joining (NJ) tree approach (Mono) showed a success rate at 100% (Fig. 1).

Barplots of measures of identification success. Abbreviations: NN, nearest-neighbour; TA, threshold analysis with 1% threshold; TA.threshVal, threshold analysis with 4.4% threshold; TA.localMinima, threshold analysis with 3.59% threshold; BCM, best close match (1% threshold); BCM.threshVal, best close match with 4.4% threshold; BCM.localMinima, threshold analysis with 3.59% threshold; Mono, proportion of monophyly on a NJ tree
Barcode gap analysis
In our reference DNA sequences, we counted how often the maximum intraspecific distance exceeded the minimum interspecific distance. Using length and which functions in SPIDER to query how many times this occurred in our reference DNA sequences, we found that this was the case on 14 occasions (Additional file 2: Figure S2).
Molecular identification for Culicoides larvae
DNA sequences of Culicoides larvae collected in the Niayes area of Senegal were successfully obtained for 958 out of 1632 larvae (58.6%). PCR amplifications failed for 99 out of 773 samples of stages L1-L2, while all selected samples of stage L3-L4 were successfully amplified (859/859 samples). This might be explained by the physical size of the different larval stages (L1 and L2 stages are < 2 mm). The sequences were edited in Geneious R11 [19] and 933 cox1 sequences of better quality were used in this study. The overall rate of cox1 sequences successfully matched within our reference DNA sequences used as Search Set in BLAST search was 97.1%. Thus, 906 out of 933 cox1 sequences of larvae were successful identified to Culicoides species. However, 27 cox1 sequences were unmatched within our DNA barcode reference libraries. In order to find a match, these cox1 sequences were used as a query in NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi). However, no matches were found for these sequences.
The sequences matched corresponded to eight Culicoides species (Table 2). Of these species, Culicoides oxystoma Kieffer had the highest percentage (66.8%), followed by Culicoides nivosus de Meillon (21.5%), Culicoides distinctipennis Austen and Culicoides similis Carter, Ingram & Macfie (both slightly above 3%) (Table 2).
DNA barcoding database analyses
A total of 1131 cox1 sequences were submitted to the BOLD database under the project code “AFCUL” (details see Additional file 3: Table S1). A hierarchical increase in mean divergence was observed according to two taxonomic levels: within species (mean = 1.92%, SE = 0.00) and within genus (mean = 17.82%, SE = 0.00). In the barcode gap analysis using the BOLD Management and Analysis System, situations where the distance to the nearest neighbour was less than the max intra-specific distance were encountered in seven species (Additional file 4: Table S2). Haplotype data analysis detected 360 haplotypes in 1131 cox1 sequences for 40 Afrotropical Culicoides species.
Discussion
Our study presents the first DNA barcode analysis of the genus Culicoides in the Afrotropical region incorporating adult and larval specimens. Biodiversity questions have become an important issue, not only in the field of conservation but also when species have an economic and health impact such as insects involved in pathogen transmission. Culicoides-borne pathogens and notably African horse sickness in the Afrotropical region are of great interest because of major outbreaks affecting horses [2,3,4,5, 20, 21]. Moreover, recent studies conducted in west and central Africa revealed high prevalence rates of Mansonella perstans both in Culicoides specimens and human populations [22,23,24].
Although of major economic and sanitary importance, the current taxonomic and ecological knowledge of Culicoides limits the understanding of the epidemiology of the diseases they transmit and therefore the implementation of appropriate and effective vector control strategies. A major limitation is that morphological methods for identifying Culicoides species are tedious and require specialized taxonomic expertise. In addition, species delimitation at the adult stage is complicated by both closely related species, for example the species of the Imicola group [18], and large morphological variations observed within certain species, in particular C. oxystoma [18]. Although morphological description and comparison of pupae of certain species has been carried out [25], especially Culicoides species related to the Similis group [26] and to the Imicola group [27, 28], there are no morphological identification keys for Culicoides larvae.
Generally, two methods have been used to identify Culicoides larvae based on the identification of emerging adults: (i) emergence traps covering potential larval habitats and allowing collection and identification of adult midges [29,30,31,32]; and (ii) collection of samples from putative breeding sites, such as mud or cattle dung, stored in laboratories for several weeks until adult midges emerge and are identified [27,28,29, 33, 34]. However, these methods are not suitable for rapid identification due to the potentially lengthy time periods of sub-adults stages, large species diversity and the maintenance efforts required to incubate samples until adult emergence. Indeed, these two methods also have bias in increasing immature mortality and therefore underestimating species diversity. In addition, adult identification problems specific to cryptic species or species with high polymorphism persist.
High-throughput identification of field-collected samples can enable insect vectors monitoring and related eco-epidemiological studies. Species identification using cox1 sequence similarity was proposed as a solution to the limitations of morphological taxonomy. The utility of DNA sequences for taxonomic or barcoding purposes is based on the nucleotide divergence [9, 35] and need critical assessment before use. cox1 barcoding sequences can be used to discover cryptic species, i.e. closely related and similar morphologically, and, for this reason, overlooked by traditional morphology-based approaches. DNA barcodes can also be used to link different life stages of insects, e.g. larvae, pupae and adults. This is particularly useful in situations where sympatry exists, or larvae are difficult to rear, as frequently occurs for Culicoides.
A first attempt to identify Culicoides larvae using molecular techniques was conducted by Yanase et al. [36] in a very restricted area in Japan and on a limited number of species. The provision of DNA barcode data for Culicoides species, particularly species of medical and veterinary importance in the Afrotropical region, fills an important gap in our knowledge of the phylogeny of these species and identification of immature Culicoides. The analysis of the quality of our DNA reference database through distance- and tree-based measures of the identification success rates showed satisfactory results (Fig. 1) and allowed its application to DNA sequences from Culicoides larvae collected in various habitats in the Niayes area of Senegal, West Africa, in order to identify species at the larval stage. The abundance of the larval stages for each species needs to be investigated in relation to the type of larval habitat sampled.
Although this study highlights that the barcode database developed here can be reliable for species-level assignments at the larval stage, the possible presence of cryptic diversity within these species is to be taken into account. Our study showed that the most abundant species in the larval sampling was C. oxystoma. Considering the vector role of C. oxystoma [37,38,39,40] its wide distribution (from Africa to South East Asia), previously described ecological heterogeneity and morphological plasticity [8, 41, 42], studies are needed to validate its taxonomic status. Culicoides oxystoma might represent a complex of species that require revision.
Of the eight Culicoides species identified at the larval stage, C. imicola is regarded as the most important vector of African horse sickness [43, 44] and bluetongue viruses [45]; C. kingi is involved in the transmission of Onchocerca gutturosa, a widespread parasite of cattle in tropical regions [46]; and C. oxystoma is a well-known vector of bovine arboviruses such as Akabane virus in Asia [37, 47]. Culicoides oxystoma and C. kingi are suspected of being vectors of African horse sickness in the Niayes region of Senegal [38, 48] based on their abundance and trophic behaviour. Larvae of C. oxystoma occupied several aquatic and semi-aquatic habitats, such as pond edge, lake edge and puddle edge in the Niayes region [33]. Larvae of this species were also found in several aquatic and semi-aquatic habitats in Japan and India, such as paddy fields, stream edges and pond margins [36, 49, 50]. In contrast, the main larval habitat of C. kingi in the Niayes region was lake edge [33]. Although adults of C. imicola can sometimes be collected in abundance in suction light traps set up at the vicinity of farms or equids in the Niayes region of Senegal [51, 52], only one cox1 sequence obtained during this study was identified as C. imicola. This confirms our previous observations that C. imicola larvae in the Niayes region have specific requirements and probably that favorable breeding sites of C. imicola have been poorly sampled or not sampled during our field investigations [33].
Conclusions
Our study provides a new diagnostic tool to help identify larvae of Culicoides at the species level in sub-Saharan Africa. These results are important regarding species of medical and veterinary interest, especially for vectors of AHSV in the Niayes area of Senegal, and serve as a point of reference for future investigations on larval ecology studies and tentative development of larval control measures that need to be selective and environmental-friendly. Besides providing reliable molecular data for species-level assignments of Afrotropical Culicoides, our study proves the efficiency of DNA barcode for studying Culicoides larval diversity from field samples. Large-scale barcode data for important taxa like Culicoides can provide a common platform to researchers from a wide array of biological studies such as taxonomy, ecology, behavior, life histories, vector control and vector-virus relationship. However, it is of prime importance that the name tagged with the generated sequences must be of high accuracy, confirmed with the expertise of a trained taxonomist, to utilize DNA barcode data for routine identification by other biologists [53]. In addition to routine identification, DNA barcode data can also provide insights into further taxonomic research through elucidation of cryptic species and resolving species complexes.
Methods
Reference DNA sequences
Reference DNA sequences constituted 230 cox1 sequences representing 42 Culicoides species (Table 1). These species were collected in different sites in the Afrotropical region [18, 41, 54, 55]. We described summary statistics and analyzed the quality of our reference DNA sequences (230 cox1 sequences representing 42 Culicoides) by distance- and tree-based measures of identification success rates using R software v.3.3.2 [56] with APE and SPIDER libraries [57, 58]. Every sequence in our reference DNA sequences was considered as unknown and used as a query against the entire data set of identified sequences, and a species name was assigned based on criteria [57]: nearest neighbour (NN), threshold analyses (TA), best close match (BCM), and monophyly of each species (Mono). These criteria are not identification tools, but permit investigation whether sequences can be used for species identification [57, 59]. The barcode gap was calculated and plotted using the maximum intraspecific distance and the minimum interspecific distance. The barcoding gap [17] is an important concept in DNA barcoding. It is assumed that the amount of genetic variation within species is smaller than the extent of variation between species. Genetic distances were calculated using SPIDER employing the Kimura-2-parameter (K2P) distance metric. Haplotype and nucleotide diversity were calculated using DnaSP v.5 [60].
Culicoides larvae sampling
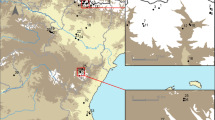
Culicoides larvae sampling was performed at four sites in the Niayes region of Senegal, West Africa: Parc de Hann, Mbao, Niague and Pout (Fig. 2). Among these, 14 larval habitats were monitored twice a month from January to December 2015, totaling 24 collection sessions. The 14 larval habitats monitored were characterized as follows: 2 larval habitats of “freshwater lake edge” in Parc de Hann (Ph1 and Ph2), 3 in Mbao (Mb1 of “pond edge”, and Mb2 and Mb3 of “saltwater lake edge”), 5 in Niague (Ng1 of “saltwater lake edge”, and Ng2, Ng3, Ng4 and Ng5 of “pond edge”), and 4 larval habitats of “puddle edge” in Pout (Pt1, Pt2, Pt3 and Pt4) (Fig. 2).

Geographical location of study sites in the Niayes region of Senegal, West Africa. Parc de Hann sites (Ph1 and Ph2) were classified as freshwater lake edge habitats. Mb1, Ng2, Ng3, Ng4 and Ng5 were pond edge while Mb2, Mb3 and Ng1were saltwater lake edge. In Pout, all habitats were puddle edge (Pt1, Pt2, Pt3 and Pt4)
For each habitat, one substrate sample of approximately 650 cm3 was collected in the upper layer of the soil surface (0–5 cm) with a trowel, filtered with a fine mesh sieve of 0.8 mm diameter and then investigated for midge larvae using a direct flotation technique in saturated sugar solution (850 g/l). Culicoides larvae were collected and preserved in 70% ethanol. A maximum of 30 individuals, irrespective of the numbers collected, were considered for molecular analyses at each of the sites sampled. If fewer than 30 individuals were collected, all individuals were analyzed (Fig. 3).

Number of Culicoides larvae collected and subsampled per site. A maximum of 30 individuals, irrespective of the numbers collected, were considered for molecular analyses at each of the sites sampled. If less than 30 individuals were collected, all individuals were analyzed
DNA extraction, polymerase chain reaction and sequencing
Genomic DNA of larvae Culicoides was individually extracted using the NucleoSpin® Tissue DNA Kit (Macherey-Nagel, Duren, Germany) according to the manufacturer’s instructions and maintained at 20 °C until further use. PCR amplification reactions were performed in a 25 μl total reaction volume containing 1× buffer, 1 mM MgCl2, 0.2 mM of each dNTP (dATP, dCTP, dGTP and dTTP), 0.2 μM forward primer LCO1490 (5'-GGT CAA CAA ATC ATA AAG ATATTG G-3'), 0.2 μM reverse primer HCO2198 (5'-TAA ACT TCA GGG TGA CCA AAA AAT CA-3') [61], 1.25 U of Taq DNA Polymerase (Qiagen, Hilden, Germany) and 0.4 ng/μl genomic DNA. The PCR cycling conditions were as follows: an initial denaturation step at 94 °C for 5 min followed by 5 cycles of 94 °C for 30 s, 45 °C for 40 s, 72 °C for 1 min, 35 cycles of 94 °C for 30 s, 51 °C for 30 s, 72 °C for 1 min, and a final extension step at 72 °C for 10 min. Positive and negative controls for the amplification reactions were carried out at every PCR round. The PCR products were separated on 1.5% agarose gels and the products were sequenced using the same primers as used in PCR amplifications (https://www.genewiz.com). All generated sequences were deposited in GenBank and BOLD.
Molecular identification for Culicoides larvae
Reference DNA sequences were transformed as a BLAST database using makeblastdb of the BLAST software v.2.2.31 [62]. To discriminate Culicoides species within the larvae generated sequences, cox1 sequences of Culicoides larvae were edited in Geneious R11 [19] and used as a query in BLAST search in the BLAST database, considering the different thresholds of divergence generated and used in the identification success rates previously described.
DNA barcoding database analyses
All DNA sequences in this study (except the sequences of C. candolfii Delécolle, Paupy, Rahola & Mathieu [54] (GenBank: KC986403.1 and KC986404.1) and C. dubitatus Kremer, Rebholtz-Hirtzel & Delécolle [55] (GenBank: KY707796.1, KY707797.1 and KY707798.1) were submitted to the BOLD database under the project code “AFCUL” for acquiring accession numbers and BOLD-IDs. Sequence alignment was performed using the BOLD Management and Analysis System [63]. Overall data sequences were compared using the Distance Summary and Barcode Gap Analysis tools on BOLD. In addition, genetic distances were calculated with the BOLD Management and Analysis System, employing the Kimura-2-parameter (K2P) distance metric [64]. Furthermore, haplotypes were calculated using DnaSP v.5 [60].
Abbreviations
- AHSV:
-
African horse sickness virus
- BCM:
-
best close match
- BOLD:
-
Barcoding of Life Database
- BTV:
-
Bluetongue virus
- EEV:
-
Equine encephalosis virus
- EHDV:
-
Epizootic hemorrhagic disease virus
- K2P:
-
Kimura 2-parameter
- Mono:
-
Monophyly
- cox1:
-
Mitochondrial gene cytochrome c oxidase subunit 1
- NJ:
-
Neighbor-joining
- NN:
-
Nearest-neighbour
- SBV:
-
Schmallenberg virus
- TA:
-
Threshold analysis
References
Borkent A. Ceratopogonidae. In: Kirk-Spriggs AH, Sinclair BJ, editors. Manual of Afrotropical Diptera, Volume 2. Nematocerous Diptera and lower Brachycera Suricata 5. Pretoria: South African National Biodiversity Institute; 2017. p. 733–812.
Purse BV, Carpenter S, Venter GJ, Bellis G, Mullens BA. Bionomics of temperate and tropical Culicoides midges: knowledge gaps and consequences for transmission of Culicoides-borne viruses. Annu Rev Entomol. 2015;60:373–92.
Carpenter S, Mellor PS, Fall AG, Garros C, Venter GJ. African horse sickness virus: history, transmission, and current status. Annu Rev Entomol. 2017;62:343–58.
Mellor PS, Boorman J, Baylis M. Culicoides biting midges: their role as arbovirus vectors. Annu Rev Entomol. 2000;45:307–40.
Akakpo AJ, Wombou Toukam CM, Mankor A, Ly C. Economic impact of African horse sickness outbreak in Senegal in 2007. Bull Anim Hlth Prod Afr. 2011;59:1–16.
Diouf ND, Etter E, Lo MM, Lo M, Akakpo AJ. Outbreaks of African horse sickness in Senegal, and methods of control of the 2007 epidemic. Vet Rec. 2012;172:152.
Meiswinkel R, Venter GJ, Nevill EM. Vectors: Culicoides spp. In: Coetzer JAW, Tustin RC, editors. Infectious diseases of livestock. Cape Town: Oxford University Press; 2004. p. 93–136.
Harrup LE, Laban S, Purse BV, Reddy YK, Reddy YN, Byregowda SM, et al. DNA barcoding and surveillance sampling strategies for Culicoides biting midges (Diptera: Ceratopogonidae) in southern India. Parasit Vectors. 2016;9:461.
Hebert PD, Cywinska A, Ball SL, deWaard JR. Biological identifications through DNA barcodes. Proc Biol Sci. 2003;270:313–21.
Meier R, Zhang G. DNA barcoding and DNA taxonomy in Diptera: an assessment based on 4261 COI sequences for 1001 species. In: Pape T, Bickel D, Meier R, editors. Diptera Diversity: Status, Challenges and Tools. Brill: Koninklijke Brill NV; 2009. p. 349–80.
Strutzenberger P, Brehm G, Fiedler K. DNA barcode sequencing from old type specimens as a tool in taxonomy: a case study in the diverse genus Eois (Lepidoptera: Geometridae). PLoS One. 2012;7:e49710.
Talaga S, Leroy C, Guidez A, Dusfour I, Girod R, Dejean A. Murienne J. DNA reference libraries of French Guianese mosquitoes for barcoding and metabarcoding. PLoS One. 2017;12:e0176993.
Tyagi K, Kumar V, Singha D, Chandra K, Laskar BA, Kundu S, et al. DNA barcoding studies on thrips in India: cryptic species and species complexes. Sci Rep. 2017;7:4898.
Besansky NJ, Severson DW. Ferdig MT. DNA barcoding of parasites and invertebrate disease vectors: what you don’t know can hurt you. Trends Parasitol. 2003;19:545–6.
Elias M, Hill RI, Willmott KR, Dasmahapatra KK, Brower AV, Mallet J, Jiggins CD. Limited performance of DNA barcoding in a diverse community of tropical butterflies. Proc Biol Sci. 2007;274:2881–9.
Avise JC. Phylogeography: the History and Formation of Species. Cambridge: Harvard University Press; 2000.
Meyer CP, Paulay G. DNA barcoding: error rates based on comprehensive sampling. PLoS Biol. 2005;3:2229–38.
Bakhoum MT, Labuschagne K, Huber K, Fall M, Mathieu B, Venter G, et al. Phylogenetic relationships and molecular delimitation of Culicoides Latreille (Diptera: Ceratopogonidae) species in the Afrotropical region: interest for the subgenus Avaritia. Syst Entomol. 2018;43:355–71.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.
Scacchia M, Lelli R, Peccio A, Di Mattia T, Mbulu RS, Hager AL, et al. African horse sickness: a description of outbreaks in Namibia. Vet Ital. 2009;45:265–74.
Venter GJ, Koekemoer JJO, Paweska JT. Investigations on outbreaks of African horse sickness in the surveillance zone in South Africa. Rev Sci Tech. 2006;25:1097–109.
Bassene H, Sambou M, Fenollar F, Clarke S, Djiba S, Mourembou G, et al. High prevalence of Mansonella perstans filariasis in rural Senegal. Am J Trop Med Hyg. 2015;93:601–6.
Debrah LB, Nausch N, Opoku VS, Owusu W, Mubarik Y, Berko DA, et al. Epidemiology of Mansonella perstans in the middle belt of Ghana. Parasit Vectors. 2017;10:15.
Simonsen PE, Onapa AW, Asio SM. Mansonella perstans filariasis in Africa. Acta Trop. 2011;120:109–20.
Nevill EM. The morphology of the immature stages of some South African Culicoides species (Diptera: Ceratopogonidae). Onderstepoort J Vet Res. 1969;36:265–84.
Nevill H, Dyce AL. Afrotropical Culicoides: description and comparison of the pupae of seven species of the Similis supergroup (Diptera: Ceratopogonidae). Onderstepoort J Vet Res. 1994;61:85–106.
Nevill H, Nevill EM, Venter GJ. Description and comparison of the pupae of a further two Culicoides (Avaritia) species from the dung of large herbivores in South Africa (Diptera: Ceratopogonidae). Onderstepoort J Vet Res. 2009;76:277–84.
Nevill H, Venter GJ, Meiswinkel R, Nevill EM. Comparative descriptions of the pupae of five species of the Culicoides imicola complex (Diptera: Ceratopogonidae) from South Africa. Onderstepoort J Vet Res. 2007;74:97–114.
Nevill EM. Biological studies on some South African Culicoides species (Diptera: Ceratopogonidae) and the morphology of their immature stages. Pretoria: University of Pretoria; 1967.
Dipeolu OO, Ogunrinade AF. Species of Culicoides breeding on rocks and riverbanks in Nigeria. Ecol Entomol. 1976;1:267–74.
Jenkins AB, Young MB. Breeding sites of Culicoides midges in KwaZulu-Natal. S Afr J Anim Sci. 2010;40:510–3.
Harrup LE, Purse BV, Golding N, Mellor PS, Carpenter S. Larval development and emergence sites of farm-associated Culicoides in the United Kingdom. Med Vet Entomol. 2013;27:441–9.
Bakhoum MT, Fall AG, Fall M, Bassene CK, Baldet T, Seck MT, et al. Insight on the larval habitat of Afrotropical Culicoides Latreille (Diptera: Ceratopogonidae) in the Niayes area of Senegal. West Africa. Parasit Vectors. 2016;9:462.
Uslu U, Dik B. Description of breeding sites of Culicoides species (Diptera: Ceratopogonidae) in Turkey. Parasite. 2007;14:173–7.
Tautz D, Arctander P, Minelli A, Thomas RH, Volger AP. A plea for DNA taxonomy. Trends Ecol Evol. 2003;18:70–4.
Yanase T, Matsumoto Y, Matsumori Y, Aizawa M, Hirata M, Kato T, et al. Molecular identification of field-collected Culicoides larvae in the southern part of Japan. J Med Entomol. 2013;50:1105–10.
Yanase T, Kato T, Kubo T, Yoshida K, Ohashi S, Yamakawa M, et al. Isolation of bovine arboviruses from Culicoides biting midges (Diptera: Ceratopogonidae) in southern Japan: 1985–2002. J Med Entomol. 2005;42:63–7.
Bakhoum MT, Fall M, Seck MT, Gardes L, Fall AG, Diop M, et al. Foraging range of arthropods with veterinary interest: new insights for Afrotropical Culicoides biting midges (Diptera: Ceratopogonidae) using the ring method. Acta Trop. 2016;157:59–67.
Fall M, Fall AG, Seck MT, Bouyer J, Diarra M, Balenghien T, et al. Circadian activity of Culicoides oxystoma (Diptera: Ceratopogonidae), potential vector of bluetongue and African horse sickness viruses in the Niayes area, Senegal. Parasitol Res. 2015;114:3151–8.
Boorman J. Culicoides (Diptera: Ceratopogonidae) of the Arabian peninsula with notes on their medical and veterinary importance. Fauna Saudi Arabia. 1989;10:160–224.
Bakhoum MT, Fall M, Fall AG, Bellis GA, Gottlieb Y, Labuschagne K, et al. First record of Culicoides oxystoma Kieffer and diversity of species within the Schultzei group of Culicoides Latreille (Diptera: Ceratopogonidae) biting midges in Senegal. PLoS One. 2013;8:e84316.
Cornet M, Brunhes J. Révision des espèces de Culicoides apparentées à C. shultzei (Enderleini, 1908) dans la région Afrotropicale (Diptera: Ceratopogonidae). Bull Soc Entomol Fr. 1994;92:149–64.
Paweska JT, Prinsloo S, Venter GJ. Oral susceptibility of South African Culicoides species to live-attenuated serotype-specific vaccine strains of African horse sickness virus (AHSV). Med Vet Entomol. 2003;17:436–47.
Venter GJ, Graham SD, Hamblin C. African horse sickness epidemiology: vector competence of south african Culicoides species for virus serotypes 3, 5 and 8. Med Vet Entomol. 2000;14:245–50.
Venter GJ, Mellor PS, Paweska JT. Oral susceptibility of South African stock-associated Culicoides species to bluetongue virus. Med Vet Entomol. 2006;20:329–34.
El Sinnary K, Hussein HS. Culicoides kingi Austen: a vector of Onchocerca gutturosa (Neumann, 1910) in the Sudan. Ann Trop Med Parasit. 1980;74:655–656.
Oem JK, Chung JY, Kwon MS, Kim TK, Lee TU, Bae YC. Abundance of biting midge species (Diptera: Ceratopogonidae, Culicoides spp.) on cattle farms in Korea. J Vet Sci. 2013;14:91–4.
Fall M, Fall AG, Seck MT, Bouyer J, Diarra M, Lancelot R, et al. Host preferences and circadian rhythm of Culicoides (Diptera: Ceratopogonidae), vectors of African horse sickness and bluetongue viruses in Senegal. Acta Trop. 2015;149:239–45.
Ray S, Choudhury A. Vertical distribution of a biting midge, Culicoides oxystoma (Diptera: Ceratopogoniadae) during different seasons in the Hooghly Estuary, Sagar Island. India. Insect Sci Appl. 1988;9:329–33.
Poddar TK, Ray S, Choudhury A. Ecology of larval Culicoides oxystoma (Diptera: Ceratopogonidea) in the Hoogly Estuary, Sagar Island India. Ann Entomol Soc Am. 1992;10:19–25.
Diarra M, Fall M, Fall AG, Diop A, Seck MT, Garros C, et al. Seasonal dynamics of Culicoides (Diptera: Ceratopogonidae) biting midges, potential vectors of African horse sickness and bluetongue viruses in the Niayes area of Senegal. Parasit Vectors. 2014;7:147.
Diarra M, Fall M, Lancelot R, Diop A, Fall AG, Dicko A, et al. Modelling the abundances of two major Culicoides (Diptera: Ceratopogonidae) species in the Niayes Area of Senegal. PLoS One. 2015;10:e0131021.
Page RDM. DNA barcoding and taxonomy: dark taxa and dark texts. Philos Trans R Soc Lond B Biol Sci. 2016;371:20150334.
Delecolle JC, Paupy C, Rahola N, Mathieu B. Morphological and molecular description of a new species of Culicoides (Avaritia) from Gabon (Diptera, Ceratopogonidae). Bull Soc Entomol Fr. 2013;118:513–9 (In French).
Augot D, Mathieu B, Hadj-Henni L, Barriel V, Zapata Mena S, Smolis S, et al. Molecular phylogeny of 42 species of Culicoides (Diptera, Ceratopogonidae) from three continents. Parasite. 2017;24:23.
R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2011.
Brown SD, Collins RA, Boyer S, Lefort MC, Malumbres-Olarte J, Vink CJ, Cruickshank RH. Spider: An R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Mol Ecol Res. 2012;12:562–5.
Paradis E, Claude J, Strimmer K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–90.
Meier R, Shiyang K, Vaidya GNP. DNA barcoding and taxonomy in Diptera: a tale of high intraspecific variability and low identification success. Syst Biol. 2006;55:715–28.
Rozas J, Sanchez-Del Barrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–7.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotech. 1994;3:294–9.
Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7:203–14.
Ratnasingham S, Hebert PDN. BOLD: The Barcode of Life Data System (http://www.barcodinglife.org). Mol Ecol Notes. 2007;7:355–64.
Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–20.
Acknowledgements
MTB would like to thank Thomas Balenghien, CIRAD, UMR ASTRE, Rabat, Morroco, for reading and commenting on the first draft. The authors are grateful to all the people who gave assistance in Culicoides collection in Senegal.
Funding
This study was funded by UMR 117 ASTRE, Cirad. MTB was funded by a grant from the Islamic Development Bank.
Availability of data and materials
All sequences generated were deposited in BOLD under the project code “AFCUL” (AFCUL001-18 to AFCUL1131-18). All data used in this study are available upon request to the corresponding author.
Author information
Authors and Affiliations
Contributions
MTB and CG designed the study. MS, KL and MF contributed the collection of Culicoides. MTB analyzed the data. KH, MC, TB and LG contributed to the manuscript after the first draft by MTB and CG. All authors read, commented on and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. The minimum cumulative error of false positive and false negative identifications show the optimum threshold; for our DNA reference libraries this was around 4.3 and 4.4%, respectively. (PDF 100 kb)
Additional file 2:
Figure S2. Line plot of the barcode gap for our DNA reference libraries. For each individual in the dataset, the light sky-blue lines represent the maximum intraspecific distance (bottom of line value), and the minimum interspecific distance (top of line value). The red lines show where this relationship is reversed, and the closest non-conspecific is actually closer to the query than its nearest conspecific, i.e. the situation where there is no barcoding gap. (PDF 3 kb)
Additional file 3:
Table S1. Details of 1131 cox1 sequences representing 40 Afrotropical Culicoides species submitted to BOLD database under the project code “AFCUL”. (XLSX 56 kb)
Additional file 4:
Table S2. Comparison between DNA sequences for Afrotropical Culicoides species using the Barcode Gap Analysis tools on BOLD. (XLSX 12 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bakhoum, M.T., Sarr, M., Fall, A.G. et al. DNA barcoding and molecular identification of field-collected Culicoides larvae in the Niayes area of Senegal. Parasites Vectors 11, 615 (2018). https://doi.org/10.1186/s13071-018-3176-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-018-3176-y