
Abstract
Background
Ticks are involved with the transmission of several viruses with significant health impact. As incidences of tick-borne viral infections are rising, several novel and divergent tick- associated viruses have recently been documented to exist and circulate worldwide. This study was performed as a cross-sectional screening for all major tick-borne viruses in several regions in Turkey. Next generation sequencing (NGS) was employed for virus genome characterization. Ticks were collected at 43 locations in 14 provinces across the Aegean, Thrace, Mediterranean, Black Sea, central, southern and eastern regions of Anatolia during 2014–2016. Following morphological identification, ticks were pooled and analysed via generic nucleic acid amplification of the viruses belonging to the genera Flavivirus, Nairovirus and Phlebovirus of the families Flaviviridae and Bunyaviridae, followed by sequencing and NGS in selected specimens.
Results
A total of 814 specimens, comprising 13 tick species, were collected and evaluated in 187 pools. Nairovirus and phlebovirus assays were positive in 6 (3.2%) and 48 (25.6%) pools. All nairovirus sequences were closely-related to the Crimean-Congo hemorrhagic fever virus (CCHFV) strain AP92 and formed a phylogenetically distinct cluster among related strains. Major portions of the CCHFV genomic segments were obtained via NGS. Phlebovirus sequencing revealed several tick-associated virus clades, including previously-characterized Antigone, Lesvos, KarMa and Bole tick viruses, as well as a novel clade. A wider host range for tick-associated virus strains has been observed. NGS provided near-complete sequences of the L genomic segments of Antigone and KarMa clades, as well as Antigone partial S segment. Co- infections of CCHFV and KarMa or novel phlebovirus clades were detected in 2.1% of the specimens.
Conclusions
Widespread circulation of various tick-associated phlebovirus clades were documented for the first time in Anatolia. Genomes of CCHFV AP92 strains were identified in previously unexplored locations. NGS provided the most detailed genomic characterization of the Antigone and KarMa viruses to date. The epidemiological and health-related consequences must be elucidated.
Similar content being viewed by others

Background
Ticks are closely-associated with the epidemiology of several microbial pathogens with significant human and animal health impact [1]. They have been implicated as arthopod vectors of many viral, bacterial, and protozoal agent and their long life-cycle, expansive range and ability to feed on a wide array of vertebrates immensely contribute to their potential for disseminating infectious agents to susceptible hosts [2]. Classified in the class Arachnida, subclass Acari, the currently-described tick species have been included in the families Argasidae (argasid or soft ticks), Ixodidae (ixodid, or hard ticks) and Nuttalliellidae [3]. Argasid and ixodid ticks can transmit a surprisingly-broad range of viruses, bacteria and protozoans, surpassing most arthropods in terms of vector potential [4].
Many tick-borne viral pathogens, causing severe morbidity and mortality, are classified in the families Flaviviridae or Bunyaviridae, such as notable human pathogens tick-borne encephalitis virus (TBEV), Crimean-Congo hemorrhagic fever virus (CCHFV), Kyasanur forest disease virus, Powassan virus, Alkhurma virus, as well as livestock pathogens African swine fever virus, Nairobi sheep disease virus, and louping ill virus [5,6,7]. Sharing the common flavivirus structure and canonical genome organization, tick-borne flaviviruses have enveloped virions with a single-stranded RNA genome in positive orientation [8]. Viral genome possesses a single open reading frame (ORF) that encodes the viral polyprotein, which requires cleavage by viral and host proteases to form the mature structural (C, preM and E), and nonstructural (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5) proteins [8]. Bunyaviruses are a diverse family of viruses with more than 400 distinct members grouped in five genera, where the major tick-borne species are classified in the genera Nairovirus and Phlebovirus [9]. All bunyaviruses are enveloped viruses with a tripartite, single-stranded RNA genome. Viral nucleocapsid protein (N), envelope glycoproteins (Gn and Gc), and RNA-dependent RNA polymerase are encoded in negative or ambisense orientation, from small (S), medium (M), and large (L) genome segments, respectively [9]. The lengths of the genomic segments vary among the genera, with the total genome lengths of approximately 11–19 kb. They also demonstrate genus-specific consensus sequences at the terminal ends of genome segments [10]. The vectors of these tick-borne infections mostly comprise ixodid ticks, classified in the genera Ixodes, Haemaphysalis, Hyalomma, Amblyomma, Dermacentor, Rhipicephalus and Boophilus [11].
Current evidence suggests a global increase in the incidence of tick-borne diseases [2]. This is not only due to an elevated frequency of endemic infections enhanced diagnosis in affected regions and expansion of tick activity zones associated with climatic factors, but to the discovery and emergence of viruses associated with ticks, as well. Several novel tick-borne viruses have been characterized within the last decade. Arguably the most prominent is the severe fever with thrombocytopenia syndrome virus (SFTSV), identified following local outbreaks of a febrile disease with high mortality rates in several provinces of China during 2009–2011 [12]. SFTSV cases or exposure has also been documented in South Korea and Japan; and Haemaphysalis longicornis ticks were suggested as the primary transmission vector [13, 14]. Another novel virus, the Heartland virus (HRTV), vectored via Amblyomma americanum ticks, has been characterized in human cases of febrile disease in the USA [15, 16]. SFTSV and HRTV are closely-related phleboviruses, and tentatively classified as a new group of tick-borne phleboviruses [17, 18]. The recent years have also witnessed the worldwide emergence of several novel tick-associated phleboviruses with unexplored human or animal health risks. Facilitated with the dissemination of next generation sequencing (NGS) technologies and application of viral metagenomics, sequences of novel strains, genetically-related to tick-borne group of phleboviruses have been characterized, from a wide range of geographical locations including the American continent [19], Africa [20, 21], Asia [22, 23], Australia [24] and Europe, around the Mediterranean region [25,26,27]. These findings clearly demonstrate the circulation of a spectrum of divergent strains in various parts of the globe and require investigation of the epidemiology as well as potential public health threats.
Located in Asia Minor and Eastern Thrace region of the Balkan Peninsula, Turkey covers a transboundary temperate climate zone between Asia and Europe. Turkey provides suitable habitats for several tick species due to the variety of ecological and climatic conditions observed throughout Anatolia [28]. Several tick-borne infections have been documented in Turkey, resulting in significant economic burden and pose major public health threats [29]. The most prominent tick-borne viral infection in Turkey is CCHF, which has emerged in 2002 and spread throughout Anatolia, with a total of 9787 cases and a mortality rate of 4.79% reported during 2002–2015 [30]. Other than CCHFV, very limited data on the presence or epidemiology of tick- borne viruses is currently available [29, 31]. This study was undertaken to perform a cross- sectional screening for all major tick-borne flaviviruses and bunyaviruses via generic PCR in several regions in Turkey, and characterize the identified strains via direct NGS on tick specimens.
Methods
Study area, specimen collection and identification
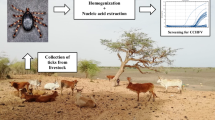
Tick sampling was carried out at 43 locations in Edirne (north-western Anatolia, Thrace region), Aydin and Mugla (western Anatolia, Aegean region), Ankara and Cankiri (central Anatolia), Sivas and Van (eastern Anatolia), Bayburt and Gumushane (northern Anatolia, Black Sea region), Antalya and Mersin (southern Anatolia, Mediterranean region), Diyarbakir, Mardin and Siirt (south-eastern Anatolia) provinces; from April to October during three consecutive years (2014–2016) (Fig. 1). At each site, flagging was performed to collect questing ticks and ticks infesting domesticated animals, mainly cattle (Bos taurus), sheep (Ovis aries), goats (Capra aegagrus hircus) and dogs (Canis familiaris) were collected at animal shelters. The specimens were kept alive in separate vials, transferred to the laboratory and identified morphologically to the species level using appropriate taxonomic keys [32,33,34,35,36]. Subsequently, ticks were pooled according to collection site, species and developmental stage up to a maximum of 20 individuals per pool and stored at -80 °C.

Illustrative map of sampling locations and distribution of the common tick species in the study
Specimen processing, nucleic acid purification and cDNA synthesis
Individual and pooled ticks with up to 5 individuals were subjected to homogenization using the SpeedMill PLUS homogenizator (Analytikjena, Jena, Germany) and subsequently, total RNA extraction via BlackPREP tick DNA/RNA kit (Analytikjena, Jena, Germany) according to the manufacturer’s instructions. Tick pools with more than 5 individuals were homogenized by vortexing with tungsten carbide or stainless steel beads (Qiagen, Hilden, Germany) in 500–700 μl of Eagle’s minimal essential medium, supplemented with 5% foetal bovine serum and 1% L-glutamine. They were clarified by centrifugation at 1430× g for 4 min, aliquoted and stored at -80 °C. Nucleic acids purification was carried out in one aliquot via High Pure Viral Nucleic Acid Kit (Roche Diagnostics, Mannheim, Germany). All specimens were subsequently subjected to complementary DNA synthesis with random hexamers, using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Hennigsdorf, Germany), performed according to the manufacturers’ guidelines.
Virus screening via generic PCR
A spectrum of assays for the detection of viruses belonging to the genera Nairovirus, Flavivirus and Phlebovirus of the families Flaviviridae and Bunyaviridae, were employed for the screening of individual or pooled ticks. Generic Nairovirus screening was carried out via a previously published single round PCR protocol with primers targeting the central motif A of the viral polymerase (on the genomic segment L) [37]. The assay could amplify 14 representative strains of all serogroups in the genus Nairovirus, including CCHFV and Nairobi sheep disease virus. Previously quantitated human plasma from CCHFV-infected individuals were employed for assay optimization [38].
A nested PCR assay with degenerated primers targeting the NS5 conserved region was used for the flavivirus screening in ticks. The assay provides a sensitive amplification of all major tick and mosquito-borne pathogenic flaviviruses including West Nile virus (WNV), dengue virus, yellow fever virus, TBEV, Murray Valley encephalitis virus, Saint Louis encephalitis virus and Usutu virus [39]. Assay optimization was accomplished via WNV NY99–4132 isolate, propagated on African green monkey (Vero) cells (ATCC-CCL81) for mosquito- borne, and TBEV strain Hypr cDNA, obtained from the European Virus Archive (http://www.european-virus-archive.com) for tick-borne strains.
Phlebovirus screening was performed using two PCR assays optimized for the amplification of tick-borne phleboviruses [20]. The binding region of the primer sets target the well-conserved viral polymerase functional motifs premotif A and motif B [40], to facilitate detection of known or novel virus strains. The assays were reported to have detection limits of 102–103 tissue culture infectious dose (TCID50) equivalents of various tick-borne phleboviruses, following RNA extraction from culture supernatants [20]. Toscana virus isolate ISS.Phl.3, propagated on Vero cells and can be robustly detected via these primer sets, was used for optimization.
Amplified products of the screening assays were visualized in a ChemiDoc XRS+ imaging system (Bio-Rad Laboratories, Munich, Germany) via ethidium bromide staining after electrophoresis in 1.3–1.7% agarose gels. Extreme care was taken to prevent carry-over contamination, and extraction, pre- and post-PCR steps were performed in spatially-separated areas.
Sequencing and phylogenetic analysis
Detectable PCR products of the screening assays were cleaned up using PureLink PCR Purification Kit (Thermo Fisher Scientific, Hennigsdorf, Germany) and characterized via sequencing in an ABI PRISM 3500xL Dx Genetic Analyzer (Thermo Fisher Scientific), using forward-reverse primers of the relevant assay and BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific). Obtained sequences were handled using CLC Main Workbench v7.8.1 (CLCBio, Aarhus, Denmark), Bioedit v.7.0.9.0 and MEGA v.6.06 softwares [41, 42].
BLASTn, BLASTn optimized for highly similar sequences (MEGABLAST) and BLASTp algorithms were employed for nucleotide similarity searches in the public databases, implemented within the National Center for Biotechnology Information website (https://blast.ncbi.nlm.nih.gov/Blast.cgi) [43]. Nucleotide and putative amino acid alignments were generated via the CLUSTAL W program, implemented within the Bioedit software [44]. Pairwise sequence comparisons were carried out via MEGA and CLC softwares. The appropriate model for the phylogenetic and molecular evolutionary analyses was determined using the Find best DNA/protein-substitution model tools built into MEGA software. Maximum likelihood trees were constructed using the Tamura-Nei and Jones-Taylor-Thornton models, based on the nucleotide and amino acid sequences, respectively. The reliability of the inferred trees was evaluated by bootstrap analysis of 1000 pseudoreplicates. Conserved protein domain and motif searches were performed using the Web CD - search tool (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) and MOTIF Search (http://www.genome.jp/tools/motif/) in the PFAM database [45, 46].
Next generation sequencing (NGS) and data analysis
Aliquoted RNA of the selected tick specimens were reverse transcribed using random hexamers into double-stranded cDNA via SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific, Hennigsdorf, Germany) and NEBNext mRNA Second Strand Synthesis Module (New England Biolabs, Frankfurt am Main, Germany). Agencourt AMPure XP Reagent (Beckman Coulter Biosciences, Krefeld, Germany) and Agilent 2100 Bioanalyzer (Agilent Technologies, Waldbronn, Germany) were used for the cleanup, yield and size distribution determination.
Fragmentation, adaptor ligation and amplification steps were performed via the NexteraXT DNA Library Preparation Kit (Illumina Inc., San Diego, CA, USA), as indicated by the manufacturer. The sequencing run was performed in one lane of the Illumina HiSeq 1500 (Illumina Inc.) in the high output mode.
The raw sequencing data was de-multiplexed and extracted in fastq format. Trimmomatic and Bowtie2 v2.2.9 softwares were employed for trimming for quality and length, removal of adaptor and background mapping [47, 48]. Acquired reads were aligned to the RefSeq viral nucleotide and protein genome database using MALT (MEGAN alignment tool, v0.3.8) and DIAMOND v0.7.1 tools [49, 50]. De novo assembly of the virus genomic segments were carried out using Geneious software v9.1 (Biomatters Ltd., Auckland, New Zealand).
Results
A total of 814 specimens, comprising 13 tick species, were collected (Additional file 1: Table S1). The specimens include 299 ticks (36.8%) from central Anatolian provinces, 255 (31.3%) from Mediterranean provinces, 191 (23.4%) from Aegean/Thrace provinces, 32 (3.9%) from eastern Anatolian provinces, 21 (2.6%) from south-eastern Anatolian provinces and 16 (1.9%) from Black Sea provinces. The most abundant tick species was Riphicephalus sanguineus complex (n = 263; 32.3%), followed by Haemaphysalis parva (n = 254; 31.2%), Riphicephalus bursa (n = 96; 11.8%) and others (Additional file 1: Table S1). The distribution of frequently- observed tick species according to the sampling province is provided in Fig. 1. Tick specimens were grouped into 187 pools, that comprise 66 pools (35.2%) from Mediterranean provinces, 49 (26.2%) from central Anatolian provinces, 27 (14.4%) from Aegean/Thrace provinces, 25 (13.3%) from eastern Anatolian provinces, 18 (9.6%) from south-eastern Anatolian provinces and 2 (1%) from Black Sea provinces (Additional file 1: Table S1).
Virus screening
A total of 50 pools (50/187, 26.7%) were positive in the screening assays, originating from central Anatolian (Cankiri: 1 pool), eastern Anatolian (Van: 12 pools), Black Sea (Bayburt: 1 pool), south-eastern Anatolian (Diyarbakir: 2 pools, Siirt: 1 pool), Mediterranean (Mersin: 28 pools) and Aegean (Mugla: 5 pools) provinces (Table 1). Generic nairovirus PCR was positive in 6 pools, collected from 3 provinces in eastern Anatolia, Black Sea and Mediterranean regions (Table 1). Positive pools comprised R. bursa (3/6) R. sanguineus complex (2/6) and Hyalomma marginatum (1/6) specimens. Generic tick phlebovirus PCR revealed positive results in 48 pools, collected from 6 provinces in eastern, south-eastern and central Anatolia, Aegean and Mediterranean regions (Table 1). Phlebovirus-positive pools comprised R. sanguineus complex (31/48), R. bursa (8/48), H. marginatum (4/48), Hyalomma aegyptium (3/48), Hyalomma excavatum (1/48) and Haemaphysalis parva (1/48) specimens. In 4 pools with R. bursa (3/4) R. sanguineus complex (1/4) specimens, nairovirus and phlebovirus co-infections were identified, with both assays providing positive results (Table 1). All specimens were negative in the generic flavivirus PCR. The contents of all reactive pools are provided in Tables 2 and 3.
Analysis and characterization of nairovirus sequences
In 6 generic nairovirus-positive specimens, sequences of 446–463 nucleotides were obtained. They demonstrated 0.9–3.0% variation and BLASTn and MEGABLAST searches revealed the most closely-related entry to be the CCHFV AP92 strain (E-value = 0.0). The sequences aligned to the 6705–7168 nucleotides on the L segment of the AP92 genome (GenBank accession number: DQ211612) and displayed 2.3–2.5% diversity from this strain. In the maximum likelihood analysis, the sequences formed a separate monophyletic cluster with the AP92 strain, distinct from other CCHFV clades, supported by high bootstrap values (Fig. 2). CCHFV sequences were detected in H. marginatum, R. sanguineus complex and R. bursa, collected from Mersin, Van and Bayburt provinces of Mediterranean and north-eastern Anatolia, in female and male individuals as well as pooled specimens (Tables 2, 3).

Maximum likelihood analysis of the partial Crimean-Congo hemorrhagic fever virus L segment sequences (445 nucleotides). The sequences characterized in this study are given in bold and indicated with a black circle, the GenBank accession number and pool code. Global virus strains are indicated by the GenBank accession number, virus and strain/isolate name. Bootstrap values higher than 50 are shown. Nairobi sheep disease virus isolate Jilin was included as the outgroup
Analysis and characterization of phlebovirus sequences
Sequencing of the generic phlebovirus amplicons provided sequences of 445–539 nucleotides with 53–100% similarity in pairwise comparisons. BLASTn and MEGABLAST analyses displayed similarities of varying degrees to several phlebovirus sequences, recently identified in Greece [25, 26], Portugal [27], USA [19] and China [23]. Maximum likelihood analysis revealed that the sequences formed distinct clades with previously-characterized tick phlebovirus sequences (Fig. 3). Four and 2 of the sequences were grouped with Antigone and Lesvos strains, identified in Greece, whereas another 8 sequences formed a distinct group with the sequences belonging in the KarMa clade, characterized in Portugal. Moreover, seven sequences grouped with the Bole tick virus, identified in China. However, the majority of the characterized sequences formed a distinct clade, which shares a common ancestor with the AnLuc lineage from Portugal (Fig. 3). Low nucleotide diversity rates were noted between sequences within the described clades; with 95.3% similarity for the Lesvos virus, 94–98.6% for KarMa virus, 90.1–99.4% for Antigone virus, 77–98% for Bole tick virus and 92.2–100% for the novel clade.

Maximum likelihood analysis of the partial tick-associated phlebovirus L segment sequences (258 nucleotides). The sequences characterized in this study are given in bold and indicated with a black diamond, the GenBank accession number and pool code. Global virus strains are indicated by the GenBank accession number, virus and strain/isolate name. Bootstrap values higher than 50 are shown
Phleboviruses Antigone, KarMa, Bole and the novel clade were detected in pools, as well as individual female and male ticks (Tables 2, 3). Association of the infection with sex could not be precisely determined for the Lesvos clade, as the sequences were detected in pools with female individuals and in mixed (Table 2). The Antigone and KarMa clades demonstrated a ubiquitous distribution, as these sequences could be detected in Van and Mugla provinces in eastern and western (Aegean) Anatolia. Similarly, Lesvos sequences were present in pools from Cankiri and Mersin provinces from central and southern (Mediterranean) Anatolia. Sequences belonging in the novel clade also originated from southern and western Anatolia. However, all specimens within the Bole clade were collected from Diyarbakir, Siirt and Van provinces of eastern and south-eastern Anatolia. This clade was also found exclusively in ticks of the genus Hyalomma. Likewise, Rhipicephalus spp. ticks were observed to harbour the Antigone clade and sequences of the novel clade was detected in R. sanguineus complex ticks (Tables 2, 3). Phlebovirus sequences could be detected in questing ticks as well as in ticks collected from vertebrate hosts.
NGS findings on phlebovirus-positive specimens
Four specimens, initially positive via the phlebovirus screening assays were evaluated via NGS and sequences of 6363–6539 nucleotides were assembled (Table 3). They demonstrated 80–99% and 87.6–99.5% similarity at the nucleotide and putative amino acid levels, respectively (Additional file 2: Table S2), and were related to the L segment of Antigone virus and other tick- borne phleboviruses in BLASTn and MEGABLAST searches. The sequences were identified to represent the Antigone (specimens MG22, MG31 and MG36) and KarMa (MG49) clades via alignment and maximum likelihood analysis with the sequence data from the screening assays (Fig. 4).

Maximum likelihood analysis of the putative, near-complete RNA-dependent RNA polymerase protein (2295 amino acids) of phleboviruses. The sequences characterized in this study are given in bold and indicated with a black triangle, the GenBank accession number and pool code. Global virus strains are indicated by the GenBank accession number, virus and strain/isolate name
In specimens MG22 and MG31, sequences of 6539 nucleotides were characterized, that represents the near-complete L segment of the Antigone clade. The putative coding regions were located identically, which encompassed the nucleotides 36–6488 on the sequence that encoded a 2161 amino acid product. Though non-coding regions of 35 and 51 nucleotides could be obtained via NGS, the sequences are considered near-complete since the phlebovirus-specific terminal motifs could be identified partially and no additional experiment was performed to ensure the availability of the complete L segment. In silico functional analyses of the polyprotein revealed several conserved motifs including the Bunyavirus N-terminus endonuclease domain (nucleotides 53–124; PSSMID: 317,864, PFAM-ID: PF15518) [51]; RNA-dependent RNA- polymerase (nucleotides 647–877 and 886–1313; PSSM-ID: 282,102, PFAM-ID: PF04196) and a viral protein of unknown function (DUF3770, nucleotides 174–426; PSSM-ID: 289,377, PFAM-ID: PF12603).
In specimens MG36 and MG49, 6374 and 6363 nucleotide-long stretches within the coding region could be obtained, which corresponded to the 2124 and 2121 amino acids of the viral RNA-dependent RNA-polymerase. The maximum likelihood analysis based on the putative amino acid sequences revealed a well-supported separation of the Antigone and KarMa clades, sharing common ancestors with Bole and Lihan tick viruses (Fig. 4). Pairwise comparison of the nucleotide and putative amino acid sequences of the L segment of tick phleboviruses with related strains are provided in Additional file 2: Table S2.
Moreover, a relatively short stretch (of 440 nucleotides) from the Antigone phlebovirus S genomic segment could be characterized via direct NGS in specimen MG22 (Table 3). BLASTn search have identified the Bole tick virus strain BL075 to be the most-closely-related strain (E- value: 2e-13) and similarity rates of 63.6 and 55.4% on the nucleotide and amino acid levels, respectively, were noted. In the maximum likelihood tree, a common ancestor of the partial Antigone S segment sequence and Bole and Lihan tick virus group were observed (Additional file 3: Figure S1). Pairwise comparison of the Antigone clade S segment sequence with related strains are provided in Additional file 4: Table S3.
NGS findings on nairovirus positive specimens
One specimen (MG48) that comprised H. marginatum specimens collected from Bayburt province of the Black Sea region was evaluated via direct NGS, following generic nairovirus PCR reactivity (Table 3). Sequences representing 3 genomic segments of the infecting nairovirus were assembled. BLASTn and MEGABLAST searches identified the CCHFV AP92 strain as the most closely-related nairovirus for all segments. Pairwise comparison of the data from each segment demonstrated that a significant portion of the coding regions and stretches of varying lengths from the 3′ non-coding ends could be characterized (Fig. 5). The strain is tentatively called as the AP92-Turkey and demonstrated 95.1, 76.2 and 90.5% nucleotide similarity of L, M and S segment sequences, respectively, with the prototype AP92 strain. Complete nucleotide and putative amino acid comparisons of viral genomic segments with various CCHFV lineages are provided in Additional files 5, 6 and 7: Tables S4-S6.

Schematic comparison and maximum likelihood analysis of the Crimean-Congo hemorrhagic fever virus L (a), M (b) and S (c) segments and coding/non-coding regions. The analysis is based on 13,125, 3928 and 1509 nucleotides for L, M and S segments, respectively. The characterized sequences are given in bold and indicated with symbols, the GenBank accession number and pool code. Global virus strains are indicated by the GenBank accession number, virus and strain/isolate name. Bootstrap values higher than 50 are provided. Nairobi sheep disease virus isolate Jilin was included as the outgroup
The obtained S segment sequence has further been aligned and analysed with previously- characterized AP92-like partial sequences from Turkey as well as several Balkan countries.
Pairwise comparison of a 426-nucleotide partial sequence revealed 98.3–99.5% similarity with sequences previously-identified in ticks (GenBank: FJ392601– FJ392603) and humans (GenBank: EU057975 and FJ392604) in Thrace region of Turkey. These sequences from Turkey and AP92-related sequences characterized from Albania, Bulgaria, Iran, Greece and Kosovo formed distinct clusters in the maximum likelihood analysis (Fig. 6).

Maximum likelihood analysis of the partial Crimean-Congo hemorrhagic fever virus S segment sequences (174 nucleotides). The AP92 and related sequences are marked. Sequences obtained in Turkey are indicated with a black square and the GenBank accession number. The sequence characterized in this study is given in bold and with the pool code. Global virus strains are indicated by the GenBank accession number, virus and strain/isolate name. Bootstrap values higher than 60 are shown. Nairobi sheep disease virus isolate Jilin was included as the outgroup
Discussion
Vector surveillance provides an efficient tool for monitoring the introduction or circulation of emerging vector-borne pathogens into susceptible populations and provide an early warning system for predicting outbreaks [52]. Furthermore, they facilitate the accumulation of information on pathogen epidemiology, associations with vectors and potential impact on human/animal welfare. Turkey provides suitable ecological niches and the tick fauna that favor the introduction and establishment of a wide range of tick-borne viral agents, as exemplified by the emergence of CCHFV [28,29,30]. This study was performed to fill the current information gap on the circulation of tick-borne viral agents via a cross-sectional vector surveillance encompassing several geographically-separated regions of Turkey. The strategy of employing generic PCR assays for a broad screening of tick-borne viruses from various families/genera and subsequent NGS was adopted, making this study the most extensive tick-borne virus surveillance effort performed in Turkey to date. During nairovirus screening, we have detected CCHFV nucleic acids in 3.2% of the specimens, collected from Mediterranean, Black Sea and eastern Anatolian provinces. All strains were genetically-related to the CCHFV AP92 strain, isolated originally from northern Greece from R. bursa ticks infecting goats in 1975 [53]. The AP92 strain is divergent from all known CCHFV strains, forming a separate clade (often referred to as the Europe 2 lineage) [54, 55]. AP92 and related strains are known to circulate in several CCHFV-endemic regions such as in the Balkan Peninsula and Iran [53,54,55,56,57,58]. In Turkey, AP92-like sequences have previously been detected in R. bursa and H. marginatum ticks, collected from Edirne and Kirklarelis provinces of the Thrace region [59]. Moreover, cases presenting as a febrile form of CCHFV disease have been described from this region, as well [60, 61]. Our findings indicate a larger dissemination zone for AP92 in Turkey, including southern and eastern Anatolia. The AP92-related sequences have also been identified in R. bursa ticks from Greece and Kosovo [54, 57], in R. sanguineus complex from Bulgaria [55] and in H. aegyptium ticks from Algeria [56]. This widespread dissemination and infrequent detection in clinical disease have led to the assumption that AP92 and related strains may be causing a milder disease in affected individuals [54]. However, a recently-documented hemorrhagic fever case with fatal outcome in Iran indicates that severe infections similar to those induced by other CCHFV lineages occur [58]. Direct NGS performed on an infected tick pool enabled the near-complete genomic characterization of the Turkish.
AP92-related strain in this study, confirming the phylogenetic relations of various CCHFV lineages and AP92 (Fig. 5). Furthermore, the analysis of partial S segment sequences has revealed distinct clusters among AP92 strains, one of which constitutes all sequences identified in Turkey (Fig. 6). However, a direct geographical association is not evident and intramural divergence within AP92-like strains related to the vector, host or the collection site needs to be addressed in future efforts.
The screening for tick-borne phleboviruses revealed a wide-spread distribution and a detection rate of 23.5%, in 7 of the 14 provinces involved and from Aegean, Mediterranean, central, eastern and south-eastern Anatolia (Table 1). Furthermore, circulation of multiple and divergent strains have been observed, with 4 previously-identified clades as well as a novel one (Fig. 3, Table 2). Currently, 3 groups of tick-associated viruses have been recognized within the genus Phlebovirus; namely the SFTSV, the Bhanja group, and the Uukuniemi groups [17, 18]. However, several recently identified sequences, as well as those reported in this study, are genetically distinct from these groups (Fig. 3). This finding indicates that only a portion of the actual tick-associated phlebovirus spectrum could be identified so far. In order to designate overall genetic relatedness and to prevent misinterpretation, we have retained the previously used nomenclature in this report [19, 23, 25,26,27]. Among the detected clades, the Antigone and Lesvos viruses have been characterized in mainland Greece and Lesvos Island in R. sanguineus complex and Hae. parva species, respectively [25, 26]. Here, we have documented a wider host range for both strains via the detection of Antigone sequences in R. bursa and Lesvos sequences in R. sanguineus complex ticks (Tables 2, 3). KarMa is among the novel phleboviruses reported from southern Portugal, along with AnLuc and RiPar clades [27]. This clade has originally been detected in R. sanguineus complex and H. marginatum ticks. Infected ticks were identified as R.bursa and H. marginatum in this study, suggesting various Rhiphicephaus ticks to be susceptible. A wide-spread distribution of Antigone, Lesvos and KarMa clades in Anatolia is likely, since they could be detected in ticks from ecologically-diverse regions.
Another phlebovirus sequence group identified in this study has been named after Bole tick virus, characterized during a metagenomic survey of the field-collected Hyalomma asiaticum ticks in China [23]. These sequences demonstrated the highest intramural nucleotide variation (up to 23%) and phylogenetic distance among the clades detected in this study (Fig. 3); thus, may be representing a group of genetically related sequences. Interestingly, the sequences could only be detected in Hyalomma spp., collected from eastern, inland regions of Anatolia (Table 2). Lastly, we have characterized several phlebovirus sequences that shared a common ancestor with the AnLuc clade from Portugal in this study [27]. We suggest these sequences to constitute a novel clade among tick-associated phleboviruses. We have refrained from specifying a tentative name, until detailed genome characterization or virus isolation has been attained.
Sequences belonging in the novel clade could be detected in R. sanguineus complex ticks from southern and western Anatolia (Table 2). Consistent with the preliminary reports from Greece, Portugal and China [23, 25,26,27], the overall phlebovirus screening findings indicate that a spectrum of viral clades circulate in various geographical regions, in questing or host-associated ticks of both sexes.
Another interesting finding of this study is the identification of phlebovirus and CCHFV co-infections in 8% (4/50) of the specimens with detectable viral nucleic acids (Table 1). The KarMa and novel clades have been observed to participate in co-infections, identified in pooled as well as in individual female and male ticks from various regions (Table 2). These findings indicate that co-infections are not extremely rare and may be contributing to virus circulation dynamics. It remains to be explored whether genetic material exchange via recombination or re- assortment, as documented for several bunya and phleboviruses [62, 63], occurs within the tick host or phlebovirus infections affect vector competence of ticks for CCHFV. Despite previous serological findings suggesting rare occurrence of tick-borne encephalitis virus exposure [29, 31], no evidence of flavivirus circulation could be identified in this study. This is presumably due to the lack of Ixodes specimens in the study cohort (Additional file 1: Table S1).
The NGS performed on selected specimens with screening reactivity have resulted in the characterization of L and S segment sequences of the Antigone and KarMa clade phleboviruses, for which only limited data have been previously available. The near-complete L segment sequences have enabled a more robust analysis of the genetic distances among partially- characterized tick-borne phleboviruses, confirming the separation of these particular clades (Fig. 4). Furthermore, this approach has provided considerable genomic sequence data of the AP92- like CCHFV strain circulating in Anatolia. As they have been instrumental in generating the information on phlebovirus diversity [19, 20, 22, 23], NGS and similar high-throughput sequencing technologies will likely to be employed for the upcoming biosurveillance efforts as well.
An important issue that remains to be elucidated is whether these identified phleboviruses are pathogenic for humans or animals or not. Among the recently-described tick-borne phleboviruses, SFTSV and Heartland virus are documented human pathogens and SFTSV is a highly-pathogenic agent causing hemorrhagic fever with a case fatality rate as high as 50% [12,13,14,15, 64]. Moreover, Uukuniemi and Bhanja viruses, the representative strains of their groups, have been associated with human febrile diseases in specific cases [65,66,67]. However, the potential virulence of other tick-borne phleboviruses, including the Antigone, Lesvos and KarMa clades detected in this study, is not currently known and they constitute potential candidates for neglected agents of febrile diseases of unknown etiology. Given the serologically-proven human and animal exposure of Uukuniemi and Bhanja viruses [68,69,70], it is likely that these clades have already been in contact with various vertebrates via ticks in endemic regions, with unexplored consequences. The current lack of an isolated strain despite previous trials [25,26,27] hampers the serological investigation of past exposures. Studies are underway by our group to inoculate the infected tick specimens onto appropriate cell lines for a successful virus isolation and detailed characterization.
Conclusion
We have documented the presence of multiple and divergent tick-associated phleboviruses throughout Anatolia, with four previously-identified as well as a novel clade. A wider host range for particular tick phlebovirus clades and a larger dissemination zone for the CCHFV AP92-like strains have been revealed. NGS has enabled the comprehensive genomic characterization of two phlebovirus clades and the CCHFV AP92-like strain in circulation.
Abbreviations
- CCHFV:
-
Crimean-Congo hemorrhagic fever virus
- DENV:
-
Dengue virus
- JEV:
-
Japanese encephalitis virus
- L:
-
Large (genomic segment)
- M:
-
Medium (genomic segment)
- NGS:
-
Next generation sequencing
- ORF:
-
Open reading frame
- RT:
-
Reverse transcription
- S:
-
Small (genomic segment)
- SFTSV:
-
Severe fever with thrombocytopenia syndrome virus;
- TBEV:
-
Tick-borne encephalitis virus
- WNV:
-
West Nile virus
- YFV:
-
Yellow fever virus
- ZIKV:
-
Zika virus
References
Dantas-Torres F, Chomel BB, Otranto D. Ticks and tick-borne diseases: a one health perspective. Trends Parasitol. 2012;28:437–46.
Kilpatrick AM, Randolph SE. Drivers, dynamics, and control of emerging vector-borne zoonotic diseases. Lancet. 2012;380:1946–55.
Guglielmone AA, Robbins RG, Apanaskevich DA, Petney TN, Estrada-Pena A, Horak IG, et al. The Argasidae, Ixodidae and Nuttalliellidae (Acari: Ixodida) of the world: a list of valid species names. Zootaxa. 2010;2538:1–28.
Jongejan F, Uilenberg G. The global importance of ticks. Parasitology. 2004;129:S3–S14.
Charrel RN, Attoui H, Butenko AM, Clegg JC, Deubel V, Frolova TV, et al. Tick-borne virus diseases of human interest in Europe. Clin Microbiol Infect. 2004;10:1040–55.
Hubalek Z, Rudolf I. Tick-borne viruses in Europe. Parasitol Res. 2012;111:9–36.
Estrada-Pena A, de la Fuente J. The ecology of ticks and epidemiology of tick-borne viral diseases. Antivir Res. 2014;108:104–28.
Simmonds P, Becher P, Collett MS, Gould EA, Heinz FX, Meyers G, et al. In: Family Flaviviridae. Virus taxonomy: classification and nomenclature of viruses. Ninth report of the international committee on taxonomy of viruses. San Diego: Elsevier; 2011. p. 1003–1020.
Plyusnin A, Beaty BJ, Elliott RM, Goldbach R, Kormelink R, et al. Bunyaviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Ninth report of the international committee on taxonomy of viruses. San Diego, CA: Elsevier Academic; 2012. p. 725–41.
Whitehouse CA, Kuhn JH, Wada J, Ergunay K. Family Bunyaviridae. In: Shapshak P, Sinnott JT, Somboonwit C, Kuhn JH, editors. Global virology I: identifying and investigating viral diseases. New York: Springer; 2015. p. 199–246.
Labuda M, Nuttall PA. Tick-borne viruses. Parasitology. 2004;129:S221–45.
Yu XJ, Liang MF, Zhang SY, Liu Y, Li JD, Sun YL, et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. New Engl J Med. 2011;364:1523–32.
Liu Q, He B, Huang SY, Wei F, Zhu XQ. Severe fever with thrombocytopenia syndrome, an emerging tick-borne zoonosis. Lancet Infect Dis. 2014;14:763–72.
Lei XY, Liu MM, Yu XJ. Severe fever with thrombocytopenia syndrome and its pathogen SFTSV. Microbes Infect. 2015;17:149–54.
McMullan LK, Folk SM, Kelly AJ, MacNeil A, Goldsmith CS, Metcalfe MG, et al. A new phlebovirus associated with severe febrile illness in Missouri. New Engl J Med. 2012;367:834–41.
Godsey MS Jr, Savage HM, Burkhalter KL, Bosco-Lauth AM, Delorey MJ. Transmission of heartland virus (Bunyaviridae: Phlebovirus) by experimentally infected Amblyomma americanum (Acari: Ixodidae). J Med Entomol. 2016;tjw080.
Matsuno K, Weisend C. Travassos Da Rosa AP, Anzick SL, Dahlstrom E, Porcella SF, et al. characterization of the Bhanja serogroup viruses (Bunyaviridae): a novel species of the genus Phlebovirus and its relationship with other emerging tick-borne phleboviruses. J Virol. 2013;87:3719–28.
Palacios G, Savji N. Travassos da Rosa AP, Guzman H, Yu X-J, Desai a, et al. characterization of the Uukuniemi virus group (Phlebovirus: Bunyaviridae): evidence for seven distinct species. J Virol. 2013;87:3187–95.
Tokarz R, Williams SH, Sameroff S, Sanchez Leon M, Jain K, Lipkin WI. Virome analysis of Amblyomma americanum, Dermacentor variabilis, and Ixodes scapularis ticks reveals novel highly divergent vertebrate and invertebrate viruses. J Virol. 2014;88:11480–92.
Matsuno K, Weisend C, Kajihara M, Matysiak C, Williamson BN, Simuunza M, et al. Comprehensive molecular detection of tick-borne phleboviruses leads to the retrospective identification of taxonomically unassigned bunyaviruses and the discovery of a novel member of the genus Phlebovirus. J Virol. 2015;89:594–604.
Kobayashi D, Ohashi M, Osei JHN, Agbosu E, Opoku M, Agbekudzi A, et al. Detection of a novel putative phlebovirus and first isolation of Dugbe virus from ticks in Accra, Ghana. Ticks Tick Borne Dis. 2017;pii: S1877-959X(16)30268–0.
Xia H, Hu C, Zhang D, Tang S, Zhang Z, Kou Z, et al. Metagenomic profile of the viral communities in Rhipicephalus spp. ticks from Yunnan, China. PLoS One. 2015;10:e0121609.
Li CX, Shi M, Tian JH, Lin XD, Kang YJ, Chen LJ, et al. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife. 2015;4 doi:10.7554/eLife.05378.
Gauci PJ, McAllister J, Mitchell IR, St George TD, Cybinski DH, Davis SS, et al. Hunter Island group Phlebovirus in ticks. Australia Emerg Infect Dis. 2015;21:2246–8.
Papa A, Kontana A, Tsioka K, Chaligiannis I, Sotiraki S. Novel phleboviruses detected in ticks. Greece. Ticks Tick Borne Dis. 2016;7:690–3.
Papa A, Kontana A, Tsioka K, Saratsis A, Sotiraki S. Novel phlebovirus detected in Haemaphysalis parva ticks in a Greek island. Ticks Tick Borne Dis. 2017;8:157–60.
Pereira A, Figueira L, Nunes M, Esteves A, Cotao AJ, et al. Multiple Phlebovirus (Bunyaviridae) genetic groups detected in Rhipicephalus, Hyalomma and Dermacentor ticks from southern Portugal. Ticks Tick Borne Dis. 2017;8:45–52.
Aydin L, Bakirci S. Geographical distribution of ticks in Turkey. Parasitol Res. 2007;101:S163–6.
Inci A, Yildirim A, Duzlu O, Doganay M, Aksoy S. Tick-borne diseases in Turkey: a review based on one health perspective. PLoS Negl Trop Dis. 2016;10:e0005021.
Leblebicioglu H, Ozaras R, Irmak H, Sencan I. Crimean-Congo hemorrhagic fever in Turkey: current status and future challenges. Antivir Res. 2016;126:21–34.
Ergunay K, Whitehouse CA, Ozkul A. Current status of human arboviral diseases in Turkey. Vector Borne Zoonotic Dis. 2011;11:731–41.
Apanaskevich DA, Horak IG. The genus Hyalomma Koch, 1844: V. Re-evaluation of the taxonomic rank of taxa comprising the H. (Euhyalomma) marginatum Koch complex of species (Acari: Ixodidae) with redescription of all parasitic stages and notes on biology. Int J Acarol. 2008;34:13–42.
Walker AR, Bouattour A, Camicas JL, Estrada-Pena A, Horak IG, Latif AA, et al. Ticks of domestic animals in Africa: a guide to identification of species. 1st ed. Bioscience reports: Edinburgh; 2003.
Filippova NA. Fauna of Russia and neighbouring countries. Ixodid ticks of subfamily Amblyomminae. Arachnoidea. 1997;4:436.
Estrada-Pena A, Bouattour A, Walker C. Ticks of domestic animals in the Mediterranean region. 1st ed. Zaragoza: University of Zaragoza Press; 2004.
Walker BJ, Kerians JE, Horak IG. The genus Rhipicephalus (Acari, Ixodidae): a guide to the Brown ticks of the world. Revised ed. Cambridge: Cambridge University Press; 2000.
Honig JE, Osborne JC, Nichol ST. The high genetic variation of viruses of the genus Nairovirus reflects the diversity of their predominant tick hosts. Virology. 2004;318:10–6.
Ergunay K, Kocak Tufan Z, Bulut C, Kinikli S, Demiroz AP, Ozkul A. Antibody responses and viral load in patients with Crimean-Congo hemorrhagic fever: a comprehensive analysis during the early stages of the infection. Diagn Microbiol Infect Dis. 2014;79:31–6.
Vazquez A, Sanchez-Seco MP, Palacios G, Molero F, Reyes N, Ruiz S, et al. Novel flaviviruses detected in different species of mosquitoes in Spain. Vector Borne Zoonotic Dis. 2012;12:223–9.
Müller R, Poch O, Delarue M, Bishop DH, Bouloy M. Rift Valley fever virus L segment: correction of the sequence and possible functional role of newly identified regions conserved in RNA-dependent polymerases. J Gen Virol. 1994;75:1345–52.
Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8.
Tamura K, Peterson D, Peterson N, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80.
Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, et al. CDD: NCBI's conserved domain database. Nucleic Acids Res. 2015;43:D222–6.
Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, et al. The Pfam protein families database. Nucleic Acids Res. 2002;30:276–80.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–9.
Huson DH, Beier S, Flade I, Gorska A, El-Hadidi M, Mitra S, et al. MEGAN community edition - interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput Biol. 2016;12:e1004957.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60.
Reguera J, Weber F, Cusack S. Bunyaviridae RNA polymerases (L-protein) have an N- terminal, influenza-like endonuclease domain, essential for viral cap-dependent transcription. PLoS Pathog. 2010;6:e1001101.
Liang G, Gao X, Gould EA. Factors responsible for the emergence of arboviruses; strategies, challenges and limitations for their control. Emerg Microbes Infect. 2015;4:e18.
Papadopoulos O, Koptopoulos G. Crimean-Congo hemorrhagic fever (CCHF) in Greece: isolation of the virus from Rhipicephalus bursa ticks and apreliminary serological survey. In: Vesenjak-Hirjan J, editor. Arboviruses in the Mediterranean countries. Stuttgart: Gustav Fischer Verlag; 1980. p. 117–21.
Papa A, Chaligiannis I, Kontana N, Sourba T, Tsioka K, Tsatsaris A, et al. A novel AP92- like Crimean-Congo hemorrhagic fever virus strain. Greece Ticks Tick Borne Dis. 2014;5:590–3.
Panayotova E, Papa A, Trifonova I, Christova I. Crimean-Congo hemorrhagic fever virus lineages Europe 1 and Europe 2 in Bulgarian ticks. Ticks Tick Borne Dis. 2016;7:1024–8.
Kautman M, Tiar G, Papa A, Siroky P. AP92-like Crimean-Congo hemorrhagic fever virus in Hyalomma aegyptium ticks. Algeria Emerg Infect Dis. 2016;22:354–6.
Sherifi K, Cadar D, Muji S, Robaj A, Ahmeti S, Jakupi X, et al. Crimean-Congo hemorrhagic fever virus clades V and VI (Europe 1 and 2) in ticks in Kosovo, 2012. PLoS Negl Trop Dis. 2014;8:e3168.
Salehi-Vaziri M, Baniasadi V, Jalali T, Mirghiasi SM, Azad-Manjiri S, Zarandi R, et al. The first fatal case of Crimean-Congo hemorrhagic fever caused by the AP92-like strain of the Crimean-Congo hemorrhagic fever virus. Jpn J Infect Dis. 2016;69:344–6.
Gargili A, Midilli K, Ergonul O, Ergin S, Alp HG, Vatansever Z, et al. Crimean-Congo hemorrhagic fever in European part of Turkey: genetic analysis of the virus strains from ticks and a seroepidemiological study in humans. Vector Borne Zoonotic Dis. 2011;11:747–52.
Midilli K, Gargili A, Ergonul O, Elevli M, Ergin S, Turan N, et al. The first clinical case due to AP92 like strain of Crimean-Congo hemorrhagic fever virus and a field survey. BMC Infect Dis. 2009;9:90.
Elevli M, Ozkul AA, Civilibal M, Midilli K, Gargili A, Duru NS. A newly identified Crimean-Congo hemorrhagic fever virus strain in Turkey. Int J Infect Dis. 2010;3:e213–6.
Briese T, Calisher CH, Higgs S. Viruses of the family Bunyaviridae: are all available isolates reassortants? Virology. 2013;446:207–16.
Lv Q, Zhang H, Tian L, Zhang R, Zhang Z, Li J, et al. Novel sub-lineages, recombinants and reassortants of severe fever with thrombocytopenia syndrome virus. Ticks Tick Borne Dis. 2017;8:385–90.
Fill MA, Compton ML, McDonald EC, Moncayo AC, Dunn JR, Schaffner W, et al. Novel clinical and pathologic findings in a heartland virus-associated death. Clin Infect Dis. 2017;64:510–2.
Calisher CH, Goodpasture HC. Human infection with Bhanja virus. Am J Trop Med Hyg. 1975;24:1040–2.
Vesenjak-Hirjan J, Calisher CH, Beus I, Marton E. First natural clinical human Bhanja virus infection. In: Vesenjak-Hirjan J, Porterfield JS, Arslanagic E, editors. Arboviruses in the Mediterranean countries: 6th FEMS symposium. Stuttgart: Fischer; 1980. p. 297–301.
Saikku P. Arboviruses in Finland. 3. Uukuniemi virus antibodies in human, cattle, and reindeer sera. Am J Trop Med Hyg. 1973;22:400–3.
Bardos V, Hubalek Z, Mittermayer T. Bhanja virus serologic survey in Czechoslovakia. Folia Parasitol. 1977;24:381.
Hubalek Z, Juricova Z. A serological survey for Bhanja virus in Czechoslovakia. Zentralbl Bakteriol Mikrobiol Hyg. 1984;258:540–3.
Hubalek Z, Mitterpak J, Prokopic J, Juricova Z, Kilik J. A serological survey for Bhanja and tick-borne encephalitis viruses in sheep of eastern Slovakia. Folia Parasitol. 1985;32:279–83.
Acknowledgements
The authors are grateful to N. Emin Güven for graphical support. Preliminary findings of this study have been submitted to the 9th Tick and Tick-borne Pathogen Conference & 1st Asia Pacific Rickettsia Conference to be held during August 27th-September 1st October, 2017, in Cairns, Australia and the 2nd International Conference on Crimean-Congo Hemorrhagic Fever to be held during September, 10–12th, 2017, in Thessaloniki, Greece.
Funding
This work has been partially supported by the Scientific and Technological Research Council of Turkey (TÜBİTAK; grant number: SBAG 115 S 074; principle investigator: AO). The corresponding author is a recipient of the Georg Forster Research Fellowship (HERMES) for Experienced Researchers by the Alexander von Humboldt Foundation, 2015. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
The nucleotide sequences characterized in this study are deposited in the GenBank database under the accession numbers KY965970–KY966011; KY979160–KY979168 and KY984485.
Author information
Authors and Affiliations
Contributions
Specimen collection and identification: ED, OH, ZK, PFP, BO and OOK; Virus screening, sequencing: ED, AB, SH, KF, PH and KE; Data interpretation: KE, AB and AÖ; Project planning, general overview, manuscript preparation: KE, AN, NÖ and AÖ. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study involved testing of host-seeking or ambusher ticks collected in the field and ticks collected from domesticated animals as specimens, and local or regional ethics committee approval was not required. Removal of ticks from infested domestic animals were performed with informed consent and cooperation of the caretakers and/or owners.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
Distribution of the tick specimens according to species, sex and collection province. (XLS 31 kb)
Additional file 2: Table S2.
Pairwise comparison of the nucleotide and putative amino acid sequences of the L segment of tick phleboviruses identified in the study (MG22, 31, 36 and 49) with related viruses. Similarity rates are given in percent. (XLS 28 kb)
Additional file 3: Figure S1.
The maximum likelihood analysis of the partial putative nucleocapsid protein (152 amino acids) of phleboviruses. The sequence characterized in this study is given in bold and indicated with the GenBank accession number, pool code and a black circle. Global virus strains are indicated by GenBank accession numbers, virus and strain/isolate names. Bootstrap values higher than 60 are shown. (PDF 422 kb)
Additional file 4: Table S3.
Pairwise comparison of the nucleotide and putative amino acid sequences of the S segment of tick phleboviruses identified in the study (MG22) with related viruses. Similarity rates are given in percent. (XLS 25 kb)
Additional file 5: Table S4.
Pairwise comparison of the nucleotide and putative amino acid sequences of the L segment of the Crimean-Congo hemorrhagic fever virus (CCHFV) identified in the study (MG48) with distinct CCHFV strains. Similarity rates are given in percent. (XLS 26 kb)
Additional file 6: Table S5.
Pairwise comparison of the nucleotide and putative amino acid sequences of the M segment of the Crimean-Congo hemorrhagic fever virus (CCHFV) identified in the study (MG48) with distinct CCHFV strains. Similarity rates are given in percent. (XLS 26 kb)
Additional file 7: Table S6.
Pairwise comparison of the nucleotide and putative amino acid sequences of the S segment of the Crimean-Congo hemorrhagic fever virus (CCHFV) identified in the study (MG48) with distinct CCHFV strains. Similarity rates are given in percent. (XLS 26 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dinçer, E., Brinkmann, A., Hekimoğlu, O. et al. Generic amplification and next generation sequencing reveal Crimean-Congo hemorrhagic fever virus AP92-like strain and distinct tick phleboviruses in Anatolia, Turkey. Parasites Vectors 10, 335 (2017). https://doi.org/10.1186/s13071-017-2279-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-017-2279-1