
Abstract
Background
Molluscs, including snails, are prone to parasite infection, which can lead to massive physiological and behavioural changes, yet many of the molecular components involved remain unresolved. Central to this point is the neural system that in snails consists of several ganglia that regulate the animals’ physiology and behaviour patterns. The availability of a genomic resource for the freshwater snail Biomphalaria glabrata provides a mean towards the high throughput analysis of changes in the central nervous system (CNS) following infection with Schistosoma miracidia.
Results
In this study, we performed a proteomic analysis of the B. glabrata CNS at pre-patent infection, providing a list of proteins that were further used within a protein-protein interaction (PPI) framework against S. mansoni proteins. A hub with most connections for both non-infected and infected Biomphalaria includes leucine aminopeptidase 2 (LAP2), which interacts with numerous miracidia proteins that together belong to the immunoglobulin family of cell adhesion related molecules. We additionally reveal the presence of at least 165 neuropeptides derived from the precursors of buccalin, enterin, FMRF, FVRI, pedal peptide 1, 2, 3 and 4, RYamide, RFamide, pleurin and others. Many of these were present at significantly reduced levels in the snail’s CNS post-infection, such as the egg laying hormone, a neuropeptide required to initiate egg laying in gastropod molluscs.
Conclusions
Our analysis demonstrates that LAP2 may be a key component that regulates parasite infection physiology, as well as establishing that parasite-induced reproductive castration may be facilitated by significant reductions in reproduction-associated neuropeptides. This work helps in our understanding of molluscan neuropeptides and further stimulates advances in parasite-host interactions.
Similar content being viewed by others


Background
The fresh water snail Biomphalaria glabrata is of medical significance as it acts as an intermediate host for the transmission of human intestinal schistosomiasis, caused by Schistosoma mansoni [1]. Schistosomiasis remains one of the most intractable public health concerns in 52 endemic countries, at least 258 million people required preventive treatment in 2014 [2]. It is second only to malaria among neglected tropical diseases in its negative impact on global human health [3] and more research is required to enable its control and eventual elimination.
Schistosoma mansoni has a complex life-cycle involving two hosts that has been reviewed extensively (e.g. [4]). One critical phase in the life-cycle requires that a miracidium, the waterborne larval stage of S. mansoni, locate and infect B. glabrata where it can initiate asexual reproduction within the host. Infected snails eventually release free-swimming cercariae that must in turn locate and infect a human host for the cycle to continue.
Sporocyst formation from miracidium within B. glabrata alters the snail’s immunity and metabolism, causing parasitic castration such that it can no longer reproduce, yet continues to support the generation of cercariae [5, 6]. Host-defence against invading miracidia appears to rely on a diverse family of fibrinogen-related proteins containing immunoglobulin-like domains [7]. Also, circulating hemocytes present in the snail hemolymph are known to encapsulate miracidia [8]. Such hemocyte-mediated cytotoxicity mechanisms include non-oxidative and oxidative pathways, involving lysosomal enzymes and reactive oxygen/nitrogen intermediates [9, 10]. An investigation of the S. mansoni miracidium proteome and its associated pathways, has provided some clues that may help to understand this host-parasite interaction [11].
Neuropeptides encompass a diverse class of chemical messengers that are instrumental in orchestrating complex physiological events from growth to reproduction and immunity. In silico data mining has revealed that at least 41 neuropeptide-like genes are present within B. glabrata, encoding precursor proteins that are predicted to release over 300 bioactive cleavage products (Adema et al., under revision). These include the neuropeptides APGWamide, conopressin, elevenin, FMRFamide, gonadotropin-releasing hormone (GnRH), whitnin, NPY and orcokinin. Although neuropeptide transcripts are represented within most of the snail tissues investigated, they are most prominent within the central nervous system (CNS) (23 of 41) and terminal genitalia (21 of 41). The achatin, GnRH, bursicon alpha, GPA2, GPB5, insulin-like peptides 1, 2 and NKY appeared to be exclusive to the snail’s CNS.
In this study, we were interested to know what molecular components were modified in the B. glabrata CNS following infection by S. mansoni miracidia. To achieve this, peptides were extracted from CNS tissue of non-infected and 12 days infected snails (pre-patent), then analysed by liquid chromatography tandem mass spectrometry (LC-MS/MS), both qualitatively and quantitatively. A protein-protein interaction network was investigated between the snail and parasite to determine putative interaction components. Then, neuropeptide abundance was assessed, with a focus on those implicated in molluscan reproduction. We additionally report the peptidomic characterisation of novel peptides displaying motifs characteristic of neuropeptides.
Results
Identification of proteins from the non-infected and infected B. glabrata CNS
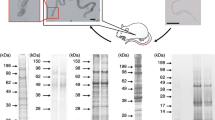
Biological triplicates of ganglia peptide extracts (non-infected and infected) were extensively fractionated by nano-HPLC, followed by tandem MS as shown in Fig. 1a. All samples were subjected to high-accuracy mass spectrometry, and the raw data were rigorously analysed using PEAKS. Using the genome [12] derived protein database (version 1.3), we identified a total of 125 peptides that matched to 68 precursor proteins (one or more unique peptides with an FDR ≤ 1%) in the non-infected B. glabrata CNS. Within the infected pre-patent snail’s CNS we identified 94 peptides from 18 precursor proteins, where only 2 were exclusive to infected CNS. There were 168 sequences supported by 257 peptides identified in non-infected snail’s CNS using the transcriptome-derived protein database, compared to 150 peptides from 69 proteins from infection samples. Fig. 1b shows the results of a comparison between the peptides identified at non-infected and pre-patent infected stages using two databases (for more details of peptide/protein identification, see Additional file 1: Table S1). The peptides and the corresponding precursor proteins only had matches in transcriptome derived protein database.

Overall workflow for identification of peptides within non-infected and infected B. glabrata. a CNS tissues were collected from three batches of snails for each state, respectively; each batch contained 12 individuals. CNS peptides were extracted using acidified methanol and extensively fractionated followed by identification with high-accuracy nano-LC TripleTOF MS. b Venn diagram comparison of peptides identified using LC-MS/MS based on genome (version 1.3) and transcriptome-derived protein databases
Protein-protein interaction (PPI) of identified B. glabrata CNS proteins with S. mansoni miracidium or sporocyst proteins
PPIs using proteins identified from the non-infected snail’s CNS with S. mansoni miracidium proteins [11] are shown in Fig. 2a. Two hubs in the PPI map from the snail cluster together, representing cytoskeletal protein spectrin and microtube actin cross-linking factor 1 (MACF1). For the PPIs of infected snail’s CNS and S. mansoni sporocyst, no MACF1 is present (Fig. 2b). One of the hubs with most connections for both non-infected and infected CNS is the leucine aminopeptidase 2 like protein (LAP2). The PPIs of the entire S. mansoni proteome versus those B. glabrata CNS proteins identified are shown in Additional file 2: Figure S1 (for protein accession names see Additional file 1: Table S1 and S. mansoni protein annotations see Additional file 3: Table S2).

Protein-protein interactions of (a) S. mansoni miracidium proteins with non-infected B. glabrata CNS proteins and (b) S. mansoni sporocyst proteins with infected CNS proteins identified. The domain-domain interaction and gene ontology annotations using DOMINE database (download on July 20th, 2014) and HMMER based on the Pfam database (version 37) were used, respectively. Each PPI with 6 or more domain-domain interaction was chosen to display, allowing for a more accurate prediction. Topological analyses were performed to explore the potential function in our constructed network using the NetworkAnalyzer plugin in Cytoscape 2.8.2
Those S. mansoni miracidium proteins that may interact with both B. glabrata LAP2 and GTP-binding proteins consist of rab and GTP-binding protein cluster. The proteins that may interact between LAP2 and fibronectin/toll-like receptor belong to a cluster of immunoglobulin family of cell adhesion related proteins, including S. mansoni hemicentin-2, roundabout 2, brother of cdo, netrin receptor dcc, titin, nephrin, receptor-type tyrosine-protein phosphatase and twitchin, while the PPIs of LAP2 and fibronectin were also detected in the infected snail’s CNS with S. mansoni sporocyst (Fig. 2b), with the S. mansoni septate junction protein additionally involved. The three hubs connecting the two major functional modules in Fig. 2a were rims binding protein 2 and tyrosine kinase (S. mansoni), and oxysterol-binding protein-related protein 9 (B. glabrata), which was missing from the PPI of infected snail’s CNS. Cytochrome c oxidase, acting as a transmembrane proton pump building an electrochemical gradient using chemical energy from the reduction of O2 [13], is another node not present (Fig. 2b). There are a total of 60 interactions that can be observed between Bg-spectrin and sporocyst proteins.
Neuropeptides identified in B. glabrata CNS by MS/MS
Numerous B. glabrata peptides identified through MS/MS did match with molluscan neuropeptide precursors. These are not represented within the PPI network since binding partners, if any, are currently unknown. Neuropeptide precursors from the B. glabrata genome were annotated with signal sequences and cleavage sites by SignalP and Neuropred, and those detected were listed in Additional file 4. Of these neuropeptide genes, our LC-MS/MS study identified 34 within non-infected snail’s CNS, and 31 within the infected snail’s CNS (Table 1). Neuropeptides that have been implicated in molluscan growth or reproduction were analysed by comparative sequence analysis and showed a precursor organisation that is consistent with homolog neuropeptides and strong amino acid conservation of the bioactive peptide regions (Fig. 3).

Schematic representation of B. glabrata neuropeptide precursors and multiple sequence alignment of bioactive peptide region with other molluscs. For each neuropeptide, a schematic precursor protein and multiple sequence alignment with other molluscs shown. Signal peptide (yellow); bioactive peptides (blue); cleavage site (vertical line); C, represents cysteine residues. In multiple sequence alignment, blue shading provides amino acid conservation. Sequence logo above alignments also show areas of high conservation
In addition to those known molluscan neuropeptides, several CNS peptides were identified that are likely processed from precursors with characteristics similar to neuropeptide precursors (i.e. signal peptide and peptide cleavage sites). The accessions of these proteins are shown in Additional file 5: Table S3 (named as Bg-NP_novel_1 to 9), supported by 23 peptides identified by LC-MS/MS. The primary sequences and MS/MS spectrum of three novel neuropeptides identified are shown in Fig. 4. Eight of the full-length sequences of these proteins were derived from the B. glabrata transcriptome (Vectorbase) and two from the genome annotation. All of these proteins were found in the non-infected CNS, and seven proteins were identified from the infected snail’s CNS, except Bg-NP_novel_7, 9 and 10 (Additional file 5: Table S3). All bioactive peptides that are predicted to be cleaved from these proteins (NeuroPred) are listed in Additional file 5: Table S3.

Sequences and MS/MS fragmentation spectra of three novel neuropeptides in B. glabrata CNS as determined by ESI-TripleTOF: Bg-NP_novel_4 - AQETINFGNQQNKPRVLA m/z 676.70, y2-y16 was detected, relatively abundant compared to the incomplete b-ion series b2-b5; Bg-NP_novel_5 - LAQMYNALKSSLKKKEPK m/z 520.06, all y-ions, b2-b4 ions were detected; and Bg-NP_novel_6 - DVDHVDLSDANDPDLLS m/z 920.40, b2, b4-b16, y13 and y16 were detected. Signal sequences (yellow), cleavage sites (red), and bioactive peptide (blue) supported by ESI-TripleTof MS/MS analysis
Differential abundance of neuropeptides in non-infected and pre-patent infected B. glabrata CNS
Potential neuropeptide precursors identified within the B. glabrata CNS were quantified using a label-free method based on the relative intensities of peptide features detected in triplicate samples identified by MS/MS. After data quality and fold change (≥ 1.5) filtration, the relative abundance of the neuropeptide precursors within infected and non-infected CNS was compared in Fig. 5. Amongst the neuropeptide precursors that were identified as differentially expressed in the B. glabrata CNS were egg laying hormone (ELH_2), whitnin (SPTR/PKYMDT/proctolin), myomodulin 2, FMRFamide, APGWamide, pedal peptide 1 (Pep 1), enterin, cerebrin, buccalin, insulin-like peptide 3, Pep 2, PTSP-like, Pep 3, Pep 4, NKY, luqin (abdominal ganglion neuropeptides L5–67), NPY, prohormone-1, AAP12, NdWF, pleurin, CCAP, sCAP 1 and 2, FVRIamide and LFRFamide. These have been identified and reviewed in several molluscan investigations [14–20]. The majority of the neuropeptides in the CNS were significantly less abundant at 12 days post-infection, such as Pep 1, Pep 2, NPY, PTSP-like, Pep 4, AAP12 and pleurin. The neuropeptide precursors that had slightly decreased included whitnin, Pep 3, NdWF, enterin, buccalin-like and CCAP. A few neuropeptides showed a relative increase in infected snail samples, including FMRFamide, luqin, NKY and sCAP 2. In addition, Bg-NP_novel_1, 2, 3, 5, 7 and 8 showed higher abundance in non-infected CNS, while there was more Bg-NP_novel_4 identified from infected CNS. Bg-NP_novel 6 was variable across biological replicates.

Neuropeptide precursor abundance in the B. glabrata CNS of non-infected versus at pre-patent stage, based on LC-MS/MS quantitative analysis. The label-free quantification module (PEAKS Q) of PEAKS v7.0 was used to compare relative concentrations of proteins at the two stages. Biological triplicates of each stage were used in tandem repeats and compared. Proteins with fold change ≥1.5 were shown (see Additional file 1: Table S1 for more details)
Additional file 6: Table S4 separates these neuropeptides by their precursor and includes those identified by LC-MS/MS. Several neuropeptides were mutually identified in both non-infected and infected snail’s CNS such as 8 buccalins, 2 ELH peptides, 8 enterins and 3 pleurins. Neuropeptides identified in non-infected CNS only, included 2 cerebrins [NGGTADTLYNIPDLMHIamide and NGGTADTLYNIPDLM(+15.99)HIa, where (+15.99) represents oxidation of methionine], ELH and its truncated form [VPIGNDLLALADLLFIERQKQAYSALKAM(+15.99)MDEAa and FIERQKQAYSALKAM(+15.99)MDEAa, respectively], 4 myomodulin peptides [PMNM(+15.99)LRLa, PMNMLRLa, SMKM(+15.99)LRLa and PM(+15.99)NMLRLa]. NPY was exclusively identified in non-infected CNS, while NKY supported by three MS peptides (GDKDDLYSAILQAAESPS, TYATTDATLETILNVLKSHAQSLRQLESTVYEQ and TYATTDATLETILNVLKSHAQSLRQLESTVYEQa) was only detected at pre-patent infection stage.
Discussion
The CNS of snails consists of several ganglia that control metabolic activities within the foot, mantle, pallial cavity, and visceral organs [21]. In this study, we provide a comprehensive analysis of the B. glabrata CNS peptides, including those that may interact with S. mansoni miracidia upon infection, as well as the neuropeptides that vary in abundance post-infection.
PPI analysis can provide valuable insight into the molecular physiological mechanisms of infection. Our identification of B. glabrata CNS proteins allowed for a semi-targeted PPI analysis with those proteins known to be present in S. mansoni. Our PPI network analysis identified the node Bg-LAP2 as the protein with highest neighbour connectivity. LAP2, like other aminopeptidases, serves vital roles in processing hormones, neurotransmission and immunological regulation, by catalysing the hydrolysis of N-terminal amino acid residues of proteins and peptides [22]. Although not characterised as LAP2, Biomphalaria seemed to have increased hemolymph levels of aminopeptidase following infection by bacteria [23] or Echinostoma lindoense [24]. In another gastropod mollusc, Aplysia, hemolymph LAP-like protein appears to degrade biologically active alpha-bag cell peptide fragments that along with the ELH peptide, regulate egg laying behaviour [25]. It has also been found that LAP2 has highest abundance in the digestive gland of mussel Mytilus edulis and its activity positively correlates with rate of whole-body protein turnover, being one of the four main lysosomal proteases associated with 73% of maintenance energy expenditure, which suggests its vital role in metabolic process [26].
Those S. mansoni miracidium proteins that cluster and interact with Bg-LAP2, fibronectin and toll-like receptor 3 include the Sm-netrin receptor, twitchin, roundabout 2, titin, nephrin and hemicentin-2-like proteins (see Fig. 2a). The interactions involving Bg-fibronectin, Sm-roundabout 2 and titin are sustained into the infection stage with sporocyst. Furthermore, there are significantly more sporocyst proteins (94) that appear to be exclusively interacting with Biomphalaria LAP2 than miracidium proteins (49), including various kinases, as well as a few receptors. Thus, it is possible that sporocysts interfere with Bg-LAP2 more intensely, thus influence the snail’s physiological activities of regulating neuropeptides for development and reproduction, or metabolism; as a result, there is likely impact on energy production pathways.
We found two hubs in the PPI map from the snail that cluster together, representing the cytoskeletal protein spectrin and MACF1. The role of MACF1 in molluscs remains unclear, although it is known to interact with actin and microtubule cytoskeletons in mammals [27, 28], where it is involved in the translocation and subsequent binding of the Axin complex to LRP6 at the cell membrane [29]. Specifically, MACF1 associates with glycogen synthase kinase 3 [30], and plays critical roles in neuronal migration in the developing brain through its interaction with the Wnt signalling pathway and control over microtubule stability [29]. It is also known that the Band 4.1-domain-containing protein (Bili) acts as a negative regulator of Wnt/β-catenin signaling due to the recruitment of Axin to LRP6 [31]. Thus, we may propose that the absence of MACF1 post-infection suggests possible inhibition of the Wnt signalling pathway, as shown in Additional file 7: Figure S2, where LRP6 phosphorylation might be blocked.
A similar scenario occurs for the oxysterol-binding protein-related protein 9 (ORP9), which was not detected post-infection. Again, its function has not been described in molluscs, yet in mice it mediates transport of sterols between the endoplasmic reticulum and trans-Golgi/TGN [32]. The absence of ORP9 within the infected snail’s CNS suggests that the transport of sterols could be suppressed. We found that ORP9 interacts with S. mansoni DDEF (development and differentiation-enhancing factor), P53 apoptosis stimulating protein, tyrosine kinase and protein kinase C (PKC). Apoptosis is known to be an important biological process that aids survival and development of Schistosoma [33], and the apoptosis-stimulating protein P53 can specifically enhance p53-induced apoptosis [34]. Sm-PKC relates directly to movement and reproduction of S. mansoni [35], while Sm-tyrosine kinases play key roles in reproduction [36].
Neuropeptides control numerous physiological activities and therefore we targeted these for quantitative analysis. Our study clearly shows that a significant modification occurs in the abundance of most B. glabrata neuropeptides at pre-patent infection stage. Several of these neuropeptides have been implicated in molluscan reproductive processes, which accords with the report that infection inhibits B. glabrata reproduction through suppression of gonad maturation [37]. One neuropeptide of significance is ELH, a well-known inducer of egg laying in the aquatic gastropod snails Aplysia and Lymnaea (called CDCH in Lymnaea, caudodorsal cell hormone) [38,39,40], and the gene has also been identified in the bivalve molluscs Crassostrea gigas and Pinctada fucata [18]. The function of Bg-ELH is still unknown, although it has enough sequence similarity compared with other gastropod ELH to suggest it may also facilitate egg laying in Biomphalaria. Thus, its down-regulation within the infected snail’s CNS would impact reproduction, which is in accordance with a recent study reporting significant impairment of B. glabrata egg laying at around seven days post-exposure to miracidia, including a decreased (P < 0.05) number of eggs per egg mass and number of eggs per snail within the first two weeks [41].
The neuropeptide, APGWa has also been implicated in reproduction. It was first discovered from the ganglia of the gastropod Fusinus ferrugineus [42] and later in other gastropods, including Lottia, Aplysia, Lymnaea and Haliotis [20, 43,44,45] and the bivalve C. gigas [18, 46]. APGWa is known to activate genital eversion in Lymnaea [47], spermiation in Helix aspersa, spawning in male Haliotis [48], and regulates egg transport and spawning in female C. gigas [46]. Whilst the oyster APGWa preprohormone contains tetrapeptides that deviate from the APGWa (i.e. KPGWa, RPGWa, SPGWa), the Bg-APGWa preprohormone contains only APGWa, a feature consistent with other gastropods [20, 44, 49].
Our analysis identified extensive peptide matches to neuropeptide families Pep 1, 2, 3 and 4 (10 to 14-amino acid peptides with a high degree of sequence similarity in each family), and similar peptides have previously been described for Aplysia [50] and Helix [51]. Peps were originally isolated from neurons ‘Pd 5’ and ‘Pd 6’, located in the pedal ganglia of the gastropod Tritonia diomedea [52], which are active during locomotion [53]. They are also found them to innovate the ciliated foot [54] where they accelerate the beat rate of ciliated foot epithelium cells [55, 56] and the epithelium of the salivary duct [57]. Significant sequence homology is observed at the C-termini of the Pep 3 sequences of gastropod species with several amino acid residues being completely conserved. The extensive sequence homology amongst the Pep 3 supports the hypothesis that they serve an important biological role and that this role is conserved.
Other neuropeptides we identified as being decreased 12 days post-infection have been associated with animal growth, including cerebrin and myomodulin in other gastropod snails. In Aplysia, cerebrin can induce arousal behaviours, similar to those obtained when offered food [58]. Moreover, Aplysia myomodulin 2 has multiple functions in behavioral plasticity, and its high expression in pedal-buccal projection neurons suggests it acts as a novel source of extrinsic modulation of the feeding system [59]. The significant down-regulation of these two neuropeptides indicates that the feeding activity of B. glabrata post-infection may be impaired. In addition, FCAP-1 and 2 were found to be less abundant in infected snails (Fig. 5). In Aplysia, FCAPs contribute to the induction and maintenance of food-induced arousal [19]. If FCAP-1, 2 plays a similar role in the B. glabrata, this would further suggest that the arousal behaviours induced by feeding in infected snails were inhibited at pre-patent stage [41].
Neuropeptides including NdWFa, AAP12 and pleurin were found to down-regulate in our study; they are known to regulate various physiological aspects in other molluscs, such as cardioexcitation [60] and participating in the calcium regulatory pathways or calcium homeostasis in terrestrial snails [61]. AAP12 has recently been associated with aestivation in the land snail Theba pisana [14]. We suggest that low-no production of these neuropeptides in Biomphalaria could have widespread implications in the snail’s functioning and biology.
Luqin, FMRFa and NKY precursors were increased at pre-patent infection stage. Luqin has been suggested to enhance tetanic contraction, phasic contraction of the Mytilus anterior byssus retractor muscle produced by repetitive electrical stimulation [62]. The tetrapeptide FMRFa is known to evoke contraction of the smooth muscles in different molluscs [63, 64]. FMRFa has also been shown to increase in the hemolymph of metabolically active Helix aspersa, leading to a decrease in glucose [65]. Thus, the increment of FMRFa-related peptides might suggest that the infected snails may become metabolically more active during this period of parasite infection.
Unlike previously reports of Pep 3, most of the novel Pep 3 identified in this study share a conserved N-terminal region of RFDRI (except RFDSISDSSAFNHFa). Conserved amino-terminal Arg-containing Pep 3 is caused by a change of a single codon in Pep 3. As shown in Additional file 6: Table S4, thirteen Pep 3 were observed, including 7 more commonly found N-terminal Pro-containing Pep 3 such as PFDRIGTSSFTSFa (m/z 730.84) and PFDRIGSSAFTSFa (m/z 715.84), and N-terminal Arg-containing Pep 3, such as RFDRIDRGSAFSRFa (m/z 432.97), RFDSISDSSAFNHFa (m/z 543.57) and RFDRISKNSQFNPFa (m/z 585.62). In addition, there was one de novo sequenced peptide RFDSISDSSAFNRFa (m/z 549.94) present in non-infected snail’s CNS also possessing the characteristics of Pep 3, with N-terminal Arg. The detection of these novel peptides provided validation of N-terminal Arg-containing sequences in Pep 3 detected in the CNS of B. glabrata.
Conclusions
We have performed an investigation of B. glabrata CNS peptides and proteins, and their association with S. mansoni infection by comparative LC-MS/MS analysis, both qualitatively and quantitatively. A PPI analysis has revealed those proteins are relevant to infection. It appears that the majority of neuropeptides analysed were significantly down-regulated post-infection, suggesting an infection-induced physiological variation in the B. glabrata CNS at pre-patent stage. We also describe the identification of a Pep 3 containing an N-terminal Arg. Our study not only significantly expands the catalogue of neuropeptides present in B. glabrata, but also provides a foundation to clarify precisely what the extracellular CNS peptides and precursor proteins are, and what roles they may play in this snail.
Methods
Animal rearing and tissue collection
The Puerto Rican strain of S. mansoni is maintained in ARC Swiss mice and B. glabrata snails at QIMR-Berghofer Medical Research Institute (QIMR-B) from stock originating from the National Institute of Allergy and Infectious Diseases Schistosomiasis Resource Centre, Biomedical Research Institute (Rockville, Maryland, USA). B. glabrata (strain BB02) were housed at QIMRB during October 2015, and maintained in flow-through aquarium tanks in a constant temperature room set to 25 °C, and fed to satiety on lettuce. For collection of CNS of ‘non-infected’ snails, animals were removed from aquaria, and were quickly killed upon removal of CNS, which was then snap frozen in liquid nitrogen. For collection of CNS of ‘infected’ snails, animals were infected with S. mansoni miracidia using standard protocols [66], the CNS was removed at 12 days post-infection and then snap frozen in liquid nitrogen. Infection was assessed by compression of tissues between two glass plates and examination under a stereoscopic microscope to confirm the presence of sporocysts. In total, CNS was collected from three batches of non-infected and infected snails, where each batch contained 12 snails (at day zero, shell diameters of 8–10 mm, and at day 12, shell diameters of 10–15 mm).
Peptide isolation and LC-MS/MS analysis of normal and infected B. glabrata CNS
The overall experimental procedure to identify and quantify CNS neuropeptides encoded within the B. glabrata genome is outlined in Fig. 1. Frozen samples of CNS were ground to a powder under liquid nitrogen in a mortar, transferred into a microfuge tube then quickly weighed in a precooled beaker. They were homogenized thoroughly in extraction buffer (90% methanol, 9% glacial acetic acid in MilliQ water) in a 1:5 w:v ratio. Crude extracts were then sonicated with three pulses, 30 s each, and centrifuged for 20 min (16,000×g, 4 °C). Supernatant was collected and lyophilised.
CNS extracts were analysed by LC-MS/MS on a Shimadzu Prominance Nano HPLC (Nakagyo-ku, Japan) coupled to a Triple Tof 5600 mass spectrometer (AB SCIEX, Concord, Canada) equipped with a nano electrospray ion source. The protocol has been detailed elsewhere [67]. Briefly, approximately 6 μl of each extract was injected and de-salted on the trap column using solvent A [0.1% formic acid (aq)] before entering a nano HPLC column (Agilent Technologies, Mulgrave, Australia) for mass spectrometry analysis. Peptide elution used a linear gradient of 1–80% solvent B [90:10 acetonitrile: 0.1% formic acid (aq)] over 120 min at 300 nl/min flow rate. Solvent B was then held at 80% for 5 min to wash the column and then returned to 1% solvent B for equilibration prior to the next sample injection. The mass spectrometer acquired 500 ms full scan TOF-MS data followed by 20 by 50 ms full scan product ion data. Full scan TOFMS data were obtained over the mass range 350–1800 and for product ion MS/MS 100–1800. Ions observed in the TOF-MS scan exceeding a threshold of 100 counts and a charge state of +2 to +5 were set to trigger the acquisition of product ion. The data were acquired and processed using Analyst TF 1.5.1 software (AB SCIEX, Concord, Canada).
Fragmentation data were analysed by PEAKS v7.0 software (BSI, Waterloo, ON, Canada). Sequences of peptides were determined by comparing the fragmentation patterns with those of proteins annotated from the B. glabrata genome (version 1.3, 14,223 entries), and also sequences derived from transcriptomes (containing 432,561 entries) within twelve tissues (https://www.vectorbase.org/). Search parameters were as follows: no enzyme was used; variable modifications included methionine oxidation, conversion of glutamine/glutamate to pyroglutamic acid, deamidation of asparagine and peptide amidation. Precursor mass error tolerance was set to 0.1 Da and a fragment ion mass error tolerance was set to 0.1 Da. De novo sequencing, database search and characterising unspecific post-translational modifications (PTMs) were used to maximise the identifications; false discovery rate (FDR) was set to ≤1%, and the individual peptide ion score [-10*Log(p)] was calculated accordingly, where p is the probability that the observed match is a random event. Proteins and their supporting peptides were obtained and analysed.
Protein-protein interaction (PPI) network
To present a PPI map, we extracted PPIs of B. glabrata proteins identified (non-infected/infected) versus S. mansoni proteins, with the miracidium proteins [11] investigated particularly, using the domain-domain interaction and gene ontology annotations. To this aim, we first adopted HMMER [68] to annotate all the known protein domains based on the Pfam database (Version 37) [69]. Using the annotated protein domain information, we used the high confidence domain-domain interaction from the DOMINE database (download on July 20th, 2014) [70] to connect those proteins with domain-domain interaction relationship. Since a protein often contains multiple domains, we required each PPI with 6 or more domain-domain interaction support to allow for a more accurate prediction. The protein database of S. mansoni was downloaded (from http://metazoa.ensembl.org/, ASM23792v2.27), from which the miracidium proteins were identified [11] and used in this study. The sporocyst protein database with full-length protein sequences was derived from its transcriptome [71], by searching the ORFs against ASM23792v2.27 using BLAST with eValue ≤10-10. In addition, topological analyses were performed to explore the potential function in our constructed network using the NetworkAnalyzer plugin in Cytoscape 2.8.2 [72]. The final network visualisation was performed using Cytoscape.
Quantitative peptide analysis
The quantitative analysis of proteins was carried out using the label-free quantification module PEAKS Q [73] of PEAKS Studio v7.0, which is based on the relative intensities of featured peptides detected in multiple samples; the detection of features was separately performed on each sample and the expectation-maximisation algorithm [74, 75] was used to identify additional overlapping features. Then, an alignment algorithm [76] was employed to align the features of the same peptide from different samples. The contents of extracted proteins in different replicate samples were quantified using a NanoDrop 2000c spectrophotometer (Thermo Scientific, Waltham, USA); for each sample, about 1.5 μg of the protein was analysed via LC-MS/MS. Biological triplicate samples of each stage (i.e. aggregation and alarm) were used in tandem repeats for LC-MS/MS procedure as described above, and the relative concentrations of proteins were compared and presented as the final results. The mass shift between different runs was set to 50 ppm, and 1.0 min was used for evaluating the retention time shift tolerance. Featured peptides with FDR threshold 1%, including PTMs mentioned above, were included in the quantitative analysis. The result of peptides was first filtrated based on: (i) ratio versus quality-score (a value indicating the quantifiable level of a peptide) and a fold change of 8 (recommendation of PEAKS Q) was used; (ii) ratio versus average-area (MS signal intensity) and set to a fold change of 8; (iii) charge of featured peptides set to between 2 and 5; (iv) fold change of peptide ≥1; and (v) featured peptide detected in more than one sample of the triplicate. Furthermore, protein results were filtered with FDR ≤ 1%, the number of unique peptides ≥1 and fold change ≥1.5. The abundance of neuropeptide precursors were compared, and proteins were clustered using one minus Pearson correlation [77].
Identification of secreted protein and neuropeptide cleavage sites
Peptide precursors were initially subject to BLASTp and tBLASTn using the corresponding database of NCBI. Protein N-terminal signal sequences were predicted using the SignalP 4.1 [78] and Predisi [79], with the transmembrane domains predicted by TMHMM [80]. For SignalP predictions, positive identifications were made when both neural network and hidden Markov model algorithms gave coincident estimations; D-cutoff values were set to 0.34 (to increase sensitivity) for both SignalP-noTM and TM networks. Herein, a protein was designated as secreted only when it met the criteria of both SignalP and Predisi and did not have a transmembrane domain predicted by TMHMM.
Biomphalaria glabrata neuropeptides were predicted by annotating protein sequences derived from the genome and transcriptome to the precursor proteins of known molluscan neuropeptides downloaded from the NCBI website (http://www.ncbi.nlm.nih.gov/). Other identified proteins were defined as novel neuropeptide-like peptides only if they were predicted to: (i) be secretory, and (ii) have multiple cleavage sites based on Neuropred [81]. De novo-only sequenced peptides were subjected to the BLAST against the neuropeptide database, NeuroPep [82], and those with matches were designated as novel neuropeptides of B. glabrata. Multiple sequence alignments were created with the Molecular Evolutionary Genetics Analysis (MEGA) software version 6.0 [83]. Sequence presentation and shading of multiple sequence alignments was performed using the LaTEX TEXshade package [84].
Abbreviations
- CNS:
-
Central nervous system
- LAP2:
-
Leucine aminopeptidase 2
- LC:
-
Liquid chromatography
- MEGA:
-
Molecular evolutionary genetics analysis
- MS/MS:
-
Mass spectrometry tandem mass spectrometry
- Pep:
-
Pedal peptide
References
Paraense WL. The schistosome vectors in the Americas. Mem Inst Oswaldo Cruz. 2001;96:7–16.
Organization WH: WHO schistosomiasis fact sheet; 2016.
King CH. Parasites and poverty: the case of schistosomiasis. Acta Trop. 2010;113(2):95–104.
Berriman M, Haas BJ, LoVerde PT, Wilson RA, Dillon GP, Cerqueira GC, et al. The genome of the blood fluke Schistosoma mansoni. Nature. 2009;460(7253):352–U65.
Xu YL, He P, Zhang L, Fang SQ, Dong SL, Zhang YJ, et al. Large-scale identification of odorant-binding proteins and chemosensory proteins from expressed sequence tags in insects. BMC Genomics. 2009;10:632.
Lafferty KD, Kuris AM. Parasitic castration: the evolution and ecology of body snatchers. Trends Parasitol. 2009;25(12):564–72.
Zhang SM, Adema CM, Kepler TB, Loker ES. Diversification of Ig superfamily genes in an invertebrate. Science. 2004;305(5681):251–4.
Lockyer AE, Spinks J, Kane RA, Hoffmann KF, Fitzpatrick JM, Rollinson D, et al. Biomphalaria glabrata Transcriptome: cDNA microarray profiling identifies resistant- and susceptible-specific gene expression in haemocytes from snail strains exposed to Schistosoma mansoni. BMC Genomics. 2008;9:634.
Bayne CJ, Hahn UK, Bender RC. Mechanisms of molluscan host resistance and of parasite strategies for survival. Parasitology. 2001;123:S159–S67.
Bayne CJ. Origins and evolutionary relationships between the innate and adaptive arms of immune systems. Integr Comp Biol. 2003;43(2):293–9.
Wang T, Zhao M, Rotgans BA, Strong A, Liang D, Ni G, et al. Proteomic analysis of the Schistosoma mansoni miracidium. PLoS One. 2016;11(1):e0147247.
Adema CM, Hillier LW, Jones CS, Loker ES, Knight M, Minx P, et al. Whole genome analysis of a schistosomiasis-transmitting freshwater snail. Nat Commun. 2017;8:15451.
Collman JP, Boulatov R, Sunderland CJ, Fu L. Functional analogues of cytochrome c oxidase, myoglobin, and hemoglobin. Chem Rev. 2004;104(2):561–88.
Adamson KJ, Wang T, Rotgans BA, Kuballa AV, Storey KB, Cummins SF. Differential peptide expression in the central nervous system of the land snail Theba pisana, between active and aestivated. Peptides. 2016;80:61–71.
Adamson KJ, Wang T, Zhao M, Bell F, Kuballa AV, Storey KB, et al. Molecular insights into land snail neuropeptides through transcriptome and comparative gene analysis. BMC Genomics. 2015;16:308.
Cropper EC, Miller MW, Tenenbaum R, Kolks M, Kupfermann I, Weiss KR. Structure and action of buccalin: a modulatory neuropeptide localized to an identified small cardioactive peptide-containing cholinergic motor neuron of Aplysia californica. Proc Natl Acad Sci USA. 1988;85(16):6177–81.
Moroz LL, Edwards JR, Puthanveettil SV, Kohn AB, Ha T, Heyland A, et al. Neuronal transcriptome of Aplysia: neuronal compartments and circuitry. Cell. 2006;127(7):1453–67.
Stewart MJ, Favrel P, Rotgans BA, Wang T, Zhao M, Sohail M, et al. Neuropeptides encoded by the genomes of the Akoya pearl oyster Pinctata fucata and Pacific oyster Crassostrea gigas: a bioinformatic and peptidomic survey. BMC Genomics. 2014;15:840.
Sweedler JV, Li L, Rubakhin SS, Alexeeva V, Dembrow NC, Dowling O, et al. Identification and characterization of the feeding circuit-activating peptides, a novel neuropeptide family of Aplysia. J Neurosci. 2002;22(17):7797–808.
Veenstra JA. Neurohormones and neuropeptides encoded by the genome of Lottia gigantea, with reference to other mollusks and insects. Gen Comp Endocrinol. 2010;167(1):86–103.
Chase R, Tolloczko B. Tracing neural pathways in snail olfaction: from the tip of the tentacles to the brain and beyond. Microsc Res Tech. 1993;24(3):214–30.
Hook V, Funkelstein L, Lu D, Bark S, Wegrzyn J, Hwang SR. Proteases for processing proneuropeptides into peptide neurotransmitters and hormones. Annu Rev Pharmacol Toxicol. 2008;48:393–423.
Cheng TC, Guida VG, Gerhart PL. Aminopeptidase and lysozyme activity levels and serum protein concentrations in Biomphalaria glabrata (Mollusca) challenged with bacteria. J Invertebr Pathol. 1978;32(3):297–302.
Cheng TC, Lie KJ, Heyneman D, Richards CS. Elevation of aminopeptidase activity in Biomphalaria glabrata (Mollusca) parasitized by Echinostoma lindoense (Trematoda). J Invertebr Pathol. 1978;31(1):57–62.
Squire CR, Talebian M, Menon JG, Dekruyff S, Lee TD, Shively JE, et al. Leucine aminopeptidase-like activity in Aplysia hemolymph rapidly degrades biologically active alpha-bag cell peptide fragments. J Biol Chem. 1991;266(33):22355–63.
Hawkins AJS, Day AJ. The metabolic basis of genetic differences in growth efficiency among marine animals. J Exp Mar Biol Ecol. 1996;203(1):93–115.
Fuchs E, Karakesisoglou I. Bridging cytoskeletal intersections. Genes Dev. 2001;15(1):1–14.
Jefferson JJ, Leung CL, Liem RK. Plakins: goliaths that link cell junctions and the cytoskeleton. Nat rev Mol Cell Biol. 2004;5(7):542–53.
Chen HJ, Lin CM, Lin CS, Perez-Olle R, Leung CL, Liem RK. The role of microtubule actin cross-linking factor 1 (MACF1) in the Wnt signaling pathway. Genes Dev. 2006;20(14):1933–45.
Ka M, Jung EM, Mueller U, Kim WY. MACF1 regulates the migration of pyramidal neurons via microtubule dynamics and GSK-3 signaling. Dev Biol. 2014;395(1):4–18.
Kategaya LS, Changkakoty B, Biechele T, Conrad WH, Kaykas A, Dasgupta R, et al. Bili inhibits Wnt/beta-catenin signaling by regulating the recruitment of axin to LRP6. PLoS One. 2009;4(7):e6129.
Ngo M, Ridgway ND. Oxysterol binding protein-related protein 9 (ORP9) is a cholesterol transfer protein that regulates Golgi structure and function. Mol Biol Cell. 2009;20(5):1388–99.
Han H, Peng J, Gobert GN, Hong Y, Zhang M, Han Y, et al. Apoptosis phenomenon in the schistosomulum and adult worm life cycle stages of Schistosoma japonicum. Parasitol Int. 2013;62(2):100–8.
Samuels-Lev Y, O'Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, et al. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell. 2001;8(4):781–94.
Ressurreicao M, De Saram P, Kirk RS, Rollinson D, Emery AM, Page NM, et al. Protein kinase C and extracellular signal-regulated kinase regulate movement, attachment, pairing and egg release in Schistosoma mansoni. PLoS Negl Trop Dis. 2014;8(6):e2924.
Morel M, Vanderstraete M, Hahnel S, Grevelding CG, Dissous C. Receptor tyrosine kinases and schistosome reproduction: new targets for chemotherapy. Front Genet. 2014;5:238.
Crews AE, Yoshino TP. Schistosoma mansoni: effect of infection on reproduction and gonadal growth in Biomphalaria glabrata. Exp Parasitol. 1989;68(3):326–34.
Strumwasser F, Schiller D, Kent S. Synthetic neuropeptide egg-laying hormone (ELH) of Aplysia californica induces normal egg-laying: structure-activity studies. Soc Neurosci Abstr. 1987;13:38.
Conn PJ, Kaczmarek LK. The bag cell neurons of Aplysia. A model for the study of the molecular mechanisms involved in the control of prolonged animal behaviors. Mol Neurobiol. 1989;3(4):237–73.
Smit A. Neuropeptide gene families that control reproductive behavior and growth in mollusks. Prog Neurobiol. 1998;54:35–54.
Faro MJ, Perazzini M, Correa Ldos R, Mello-Silva CC, Pinheiro J, Mota EM, et al. Biological, biochemical and histopathological features related to parasitic castration of Biomphalaria glabrata infected by Schistosoma mansoni. Exp Parasitol. 2013;134(2):228–34.
Kuroki Y, Kanda T, Kubota I, Fujisawa Y, Ikeda T, Miura A, et al. A molluscan neuropeptide related to the crustacean hormone, RPCH. Biochem. Biophys Res Commun. 1990;167(1):273–9.
Smit A, Jiménez C, Dirks R, Croll R, Geraerts W. Characterization of a cDNA clone encoding multiple copies of the neuropeptide APGWamide in the mollusk Lymnaea stagnalis. J Neurosci. 1992;12(5):1709–15.
Fan X, Croll RP, Wu B, Fang L, Shen Q, Painter SD, et al. Molecular cloning of a cDNA encoding the neuropeptides APGWamide and cerebral peptide 1: localization of APGWamide like immunoreactivity in the central nervous system and male reproductive organs of Aplysia. J Comp Neurol. 1997;387(1):53–62.
Chansela P, Saitongdee P, Stewart P, Soonklang N, Stewart M, Suphamungmee W, et al. Existence of APGWamide in the testis and its induction of spermiation in Haliotis asinina Linnaeus. Aquaculture. 2008;279(1–4):142–9.
Bernay B, Baudy-Floc'h M, Zanuttini B, Zatylny C, Pouvreau S, Henry J. Ovarian and sperm regulatory peptides regulate ovulation in the oyster Crassostrea gigas. Mol Reprod dev. 2006;73(5):607–16.
Koene JM. Neuro-endocrine control of reproduction in hermaphroditic freshwater snails: mechanisms and evolution. Front Behav Neurosci. 2010;4:167.
Chansela P, Saitongdee P, Stewart P, Soonklang N, Hanna PJ, Nuurai P, et al. The tetrapeptide APGW-amide induces somatic growth in Haliotis asinina Linnaeus. J Shellfish Res. 2010;29 3:753–6.
York PS, Cummins SF, Degnan SM, Woodcroft BJ, Degnan BM. Marked changes in neuropeptide expression accompany broadcast spawnings in the gastropod, Haliotis asinina. Front Zool. 2012;9:1–9.
Lloyd PE, Connolly CM. Sequence of pedal peptide: a novel neuropeptide from the central nervous system of Aplysia. J Neurosci. 1989;9(1):312–7.
Poteryaev DA, Zakharov IS, Balaban PM, Uvarov PN, Belyavsky AV. Characterization of a cDNA clone encoding pedal peptide in the terrestrial snail. Neuroreport. 1997;8(16):3631–5.
Lloyd PE, Phares GA, Phillips NE, Willows AO. Purification and sequencing of neuropeptides from identified neurons in the marine mollusc, Tritonia. Peptides. 1996;17(1):17–23.
Popescu IR, Willows AO. Sources of magnetic sensory input to identified neurons active during crawling in the marine mollusc Tritonia diomedea. J exp Biol. 1999;202(21):3029–36.
Willows A, Dorsett D, Hoyle G. The neuronal basis of behavior in Tritonia. III. Neuronal mechanism of a fixed action pattern. J Neurobiol. 1973;4(3):255–85.
Willows AO, Pavlova GA, Phillips NE. Modulation of ciliary beat frequency by neuropeptides from identified molluscan neurons. J Exp Biol. 1997;200(10):1433–9.
Willows AO, Pavlova GA, Phillips NE. Effect of Tritonia neuropeptides and serotonin on ciliary activity. Dokl Akad Nauk. 1998;358(6):839–41.
Gaston MR. Neuropeptide TPep action on salivary duct ciliary beating rate in the nudibranch mollusc Tritonia diomedea. Invertebr Neurosci. 1998;3(4):327–33.
Li L, Floyd PD, Rubakhin SS, Romanova EV, Jing J, Alexeeva VY, et al. Cerebrin prohormone processing, distribution and action in Aplysia californica. J Neurochem. 2001;77(6):1569–80.
Proekt A, Vilim FS, Alexeeva V, Brezina V, Friedman A, Jing J, et al. Identification of a new neuropeptide precursor reveals a novel source of extrinsic modulation in the feeding system of Aplysia. J Neurosci. 2005;25(42):9637–48.
Morishita F, Nakanishi Y, Sasaki K, Kanemaru K, Furukawa Y, Matsushima O. Distribution of the Aplysia cardioexcitatory peptide, NdWFamide, in the central and peripheral nervous systems of Aplysia. Cell Tissue Res. 2003;312(1):95–111.
Bogdanov YD, Balaban PM, Poteryaev DA, Zakharov IS, Belyavsky AV. Putative neuropeptides and an EF-hand motif region are encoded by a novel gene expressed in the four giant interneurons of the terrestrial snail. Neurosci. 1998;85(2):637–47.
Fujimoto K, Ohta N, Yoshida M, Kubota I, Muneoka Y, Kobayashi M. A novel cardio-excitatory peptide isolated from the atria of the African giant snail, Achatina fulica. Biochem Biophys Res Commun. 1990;167(2):777–83.
Price DA, Greenberg MJ. Pharmacology of the molluscan cardioexcitatory neuropeptide FMRFamide. Gen Pharmacol-Vasc S. 1980;11(2):237–41.
Nelson ID, Huddart H. Neuromodulation in molluscan smooth muscle: the action of 5-HT, FMRFamide and purine compounds. Gen Pharmacol. 1994;25(3):539–52.
Roszer T, Kiss-Toth ED. FMRF-amide is a glucose-lowering hormone in the snail Helix aspersa. Cell Tissue Res. 2014;358(2):371–83.
Tucker MS, Karunaratne LB, Lewis FA, Freitas TC, Liang YS. Schistosomiasis. In: Coico R, editor. Current Protocols in Immunology 19.1.1-19.1.57. John Wiley and Sons, Inc. Published online November 2013 in Wiley Online Library (wileyonlinelibrary.com) doi:10.1002/0471142735.im1901s103.
Hall MR, Kocot KM, Baughman KW, Fernandez-Valverde SL, Gauthier MEA, Hatleberg WL, et al. The crown-of-thorns starfish genome as a guide for biocontrol of this coral reef pest. Nature. 2017;544 7649:231–4.
Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(Web Server issue):W29–37.
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42(Database issue):D222–30.
Yellaboina S, Tasneem A, Zaykin DV, Raghavachari B, Jothi R. DOMINE: a comprehensive collection of known and predicted domain-domain interactions. Nucleic Acids Res. 2011;39(Database issue):D730–5.
Vermeire JJ, Taft AS, Hoffmann KF, Fitzpatrick JM, Yoshino TP. Schistosoma mansoni: DNA microarray gene expression profiling during the miracidium-to-mother sporocyst transformation. Mol Biochem Parasitol. 2006;147(1):39–47.
Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431–2.
Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.P.B.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnol. 2008;26(12):1367–72.
Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75(17):4646–58.
Nesvizhskii AI, Vitek O, Aebersold R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nature Med. 2007;4(10):787–97.
Lin H, He L, Ma B. A combinatorial approach to the peptide feature matching problem for label-free quantification. Bioinformatics. 2013;29(14):1768–75.
Lin LI. A concordance correlation coefficient to evaluate reproducibility. Biometrics. 1989;45(1):255–68.
Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–6.
Hiller K, Grote A, Scheer M, Munch R, Jahn D. PrediSi: prediction of signal peptides and their cleavage positions. Nucleic Acids Res. 2004;32:W375–9.
Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–80.
Southey BR, Amare A, Zimmerman TA, Rodriguez-Zas SL, Sweedler JV. NeuroPred: a tool to predict cleavage sites in neuropeptide precursors and provide the masses of the resulting peptides. Nucleic Acids Res. 2006;34:W267–72.
Wang Y, Wang M, Yin S, Jang R, Wang J, Xue Z, et al. NeuroPep: a comprehensive resource of neuropeptides. Database. 2015;2015:bav038.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Beitz E. TEXshade: shading and labeling of multiple sequence alignments using LATEX2 epsilon. Bioinformatics. 2000;16(2):135–9.
Acknowledgements
We acknowledge the help of Ms. Mary Duke of QIMR Berghofer with the egg hatching and the miracidia processing and harvesting. We thank Prof. Karl Hoffmann (the Animal & Microbial Sciences Research Theme, Aberystwyth University, UK) for providing the transcriptome data of S. mansoni sporocyst, and Dr. Alun Jones (Institute for Molecular Bioscience, the University of Queensland) for advice and assistance with the LC-MS/MS. B. glabrata snails provided by the NIAID Schistosomiasis Resource Center of the Biomedical Research Institute (Rockville, MD) through NIH-NIAID Contract HHSN272201000005I for distribution through BEI Resources. This research was undertaken with a generous computing time allocation from the National Computational Infrastructure (NCI), which is supported by the Australian Government.
Availability of data and materials
The original S. mansoni protein dataset is available from the GeneDB (www.genedb.org) and SchistoDB (www.schistodb.net) databases.
Funding
We thank the University of the Sunshine Coast who provided an internal grant to help support this work (TW). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authors’ contributions
Conceived and designed the study and supervised the project: SFC and TW. Undertook the study and data analysis: DL, MZ and TW. Contributed to analysis using various tools: DL, MZ, UB, TW and SFC. Wrote the paper: SK, DPM and SFC. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Schistosoma mansoni is maintained at the QIMR Berghofer Medical Research (QIMR-B) Institute under permit from the Australian Department Agriculture, Fisheries and Forestry Biosecurity (DAFF). This study was performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The conduct and procedures involving animal experimentation were approved by the Animal Ethics Committee of QIMR-B (project number P242).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1: Table S1.
Identification and quantification of neuropeptide precursor proteins from non-infected/S. mansoni-infected CNS of B. glabrata; the identification of genome or transcriptome-derived proteins and protein annotations. (XLSX 197 kb)
Additional file 2: Figure S1.
Protein-protein interactions between proteins identified in non-/infected B. glabrata CNS and S. mansoni. (TIFF 1844 kb)
Additional file 3: Table S2.
Proteins on the PPI network and the annotations of relevant proteins of S. mansoni. (XLSX 173 kb)
Additional file 4: File S1.
List of neuropeptide precursors annotated with signal sequences (predicted by SignalP 4.1, yellow) and cleavage sites (predicted by NeuroPrep, red), supported by ESI-TripleTof MS/MS analysis of B. glabrata CNS. Cystine residues are shaded green. (DOCX 19 kb)
Additional file 5: Table S3.
Sequences of the precursors of novel neuropeptides, peptides identified by LC-MS/MS and bioactive peptides cleaved from the precursors predicted by Neuropred. (XLSX 19 kb)
Additional file 6: Table S4.
Neuropeptide matches identified in the CNS of non-infected (NInf) and S. mansoni-infected (Inf) Biomphalaria glabrata by LC-MS/MS. Lower case “a” at the C-terminal peptide indicates C-terminal amidation. (XLSX 18 kb)
Additional file 7: Figure S2.
Wnt/β-catenin signalling pathway, the recruitment of Axin–GSK3β to the membrane is regulated positively by MACF1 and negatively by Bili, whereas Caprin-2 stabilises the LRP6 and Axin–GSK3β complex, and MAPKs or GRK5/6 phosphorylates the PPPS/TP motifs of LRP6. S. mansoni infection eliminates MACF1 from B. glabrata, thereby blocking the signal. (TIFF 1206 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, T., Zhao, M., Liang, D. et al. Changes in the neuropeptide content of Biomphalaria ganglia nervous system following Schistosoma infection. Parasites Vectors 10, 275 (2017). https://doi.org/10.1186/s13071-017-2218-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-017-2218-1