
Abstract
Background
Enolase is an essential multifunctional glycolytic enzyme that is involved in many biological processes of apicomplexan protozoa, such as adhesion and invasion. However, the characteristics of enolase in Cryptosporidium parvum, including the location on the oocyst and the enzyme activity, remain unclear.
Methods
The C. parvum enolase gene (cpeno) was amplified by RT-PCR and sequenced. The deduced amino acid sequence was analysed by bioinformatics software. The gene was expressed in Escherichia coli BL21 (DE3) and purified recombinant protein was used for enzyme activity analysis, binding experiments and antibody preparation. The localisation of enolase on oocysts was examined via immunofluorescence techniques.
Results
A 1,350 bp DNA sequence was amplified from cDNA taken from C. parvum oocysts. The deduced amino acids sequence of C. parvum enolase (CpEno) had 82.1% homology with Cryptosporidium muris enolase, and 54.7–68.0% homology with others selected species. Western blot analysis indicated that recombinant C. parvum enolase (rCpEno) could be recognised by C. parvum-infected cattle sera. Immunolocalization testing showed that CpEno was found to locate mainly on the surface of oocysts. The enzyme activity was 33.5 U/mg, and the Michaelis constant (K m ) was 0.571 mM/l. Kinetic measurements revealed that the most suitable pH value was 7.0–7.5, and there were only minor effects on the activity of rCpEno with a change in the reaction temperature. The enzyme activity decreased when the Ca2+, K+, Mg2+ and Na+ concentrations of the reaction solution increased. The binding assays demonstrated that rCpEno could bind to human plasminogen.
Conclusion
This study is the first report of immunolocation, binding activity and enzyme characteristics of CpEno. The results of this study suggest that the surface-associated CpEno not only functions as a glycolytic enzyme but may also participate in attachment and invasion process of the parasite.
Similar content being viewed by others


Background
Cryptosporidium is an important zoonotic protozoan with a wide range of hosts, including humans and various animals. The main symptoms of cryptosporidiosis are diarrhoea, which can be fatal [1]. Cryptosporidium is the second most serious diarrheal disease in infants and young children in developing countries after rotavirus [2]. According to the World Health Organization, there are nearly eight million annual deaths of children under 5 years of age caused by diarrhoea [3].
Currently, no effective vaccine or drug has been found to prevent this disease, so it is important to identify specific target antigens and better understand the host immune response to the parasite. Nowadays, many moonlighting proteins in Cryptosporidium have been found to play an important role in parasite adhesion and invasion. For example, C. parvum elongation factor 1α (EF-1α), a novel protein associated with host cell invasion could be a candidate vaccine antigen against cryptosporidiosis [4]. Cryptosporidium parvum Clec (CpClec), a novel mucin-like glycoprotein, contains a C-type lectin domain (CTLD) and localised to the surface of the apical region, maybe playing a significant role in Cryptosporidium-host cell interactions [5].
Unlike other apicomplexan parasites, such as Plasmodium falciparum and Toxoplasma gondii, Cryptosporidium lacks many of the pharmacological targets, and no mitochondrial genome has been found [6]. Energy metabolism is a necessary process of biological survival, and the main way to obtain energy in the majority of higher organisms is the tricarboxylic acid (TCA) cycle and β-oxidation process [7]. However, according to the complete genome sequence, these metabolic pathways are absent in C. parvum [8]. Therefore, the main energy pathway in C. parvum is probably glycolysis [9], so enzymes involved in the glycolytic pathway may be potential targets for therapeutic agents.
As a key enzyme in the glycolytic pathway, enolase can catalyse the reversible interconversion of 2-phosphoglycerate (2-PGA) and phosphoenolpyruvate (PEP), which exist in many species. Many studies have clarified that enolase is a highly conserved and multifunctional protein in both prokaryotes and eukaryotes [10]. It has been localised in the cytoplasm, cell surface and nucleus of various mammalian cells [11]. According to previous reports, enolase has many moonlighting functions [12,13,14,15,16,17,18,19]. For example, enolase can be displayed on the surface of several kinds of cells including certain tumour cells and act as a plasminogen binding receptor, which promotes tissue invasion and dissemination through the body [12,13,14,15,16,17]. Enolase in the RNA degradosome plays a crucial role in the rapid decay of glucose transporter mRNA in response to phosphosugar stress in Escherichia coli [18]. In Arabidopsis thaliana, enolase can bind to the zinc finger protein gene and impair cold-responsive gene transcription [19]. Nuclear regulatory functions exist in the T. gondii enolase that may involve changes in chromatin structure [20]. In P. falciparum, biliverdin (BV) can target enolase and eukaryotic initiation factor (eIF2α) to reduce the intraerythrocytic development of the parasite [21]. In Cryptosporidium spp., several sequences of enolase genes have been published, including that for C. hominis (GenBank XM_663094), C. muris (GenBank NW_002196570) and C. parvum (GenBank XM_626138). However, to our knowledge, until now there was no related report concerning the characteristics and functions of enolase in Cryptosporidium spp. The aim of the present study was to investigate the localisation, binding activity, enzymatic activity and factors that may influence the activity of enolase in C. parvum.
Methods
Parasite multiplication and purification
The Iowa isolate oocysts of C. parvum were purchased from Waterborne Inc. (New Orleans, LA, USA) and multiplied in newborn Holstein bull calves in our laboratory. Oocysts from the faeces were concentrated with Sheather’s sucrose flotation as previously described by Chen et al. [22] and purified by discontinuous sucrose gradients [23]. All the purified oocysts were stored in 2.5% potassium dichromate (K2Cr2O7) solution at 4 °C and used for RNA extraction or immunofluorescence assay within 1 month.
Gene amplification, sequence and phylogenetic analysis
Total RNA was extracted from freshly isolated oocysts of C. parvum using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA), and precipitated with isopropanol, rinsed with 70% ethanol, then finally dissolved in 20 μl of RNase-free water. RNA integrity was checked in 1.0% agarose gels and concentration was tested by NanoDrop 2000C (Thermo Fisher Scientific, Carlsbad, CA, USA) by reading extinction values at 260/280 nm. The total RNA was reversely transcribed into cDNA by a PrimeScript™ II Reverse Transcriptase kit according to the manufacture’s protocol (Takara, Dalian, China).
The C. parvum enolase (CpEno) encoding gene (cpeno) was amplified by polymerase chain reaction (PCR) using a forward primer 5′-GGA ATT CAG TTT AGC CAT GCC TTC TAT ATT GTC-3′ and the reverse primer 5′-CCC AAG CTT TTA TCT GAT TCT TGC GAT GGG TTT TC-3′ which introduced restriction endonuclease sites EcoR I and Hind III (underlined), respectively. PCR was performed using the following conditions: an initial denaturation at 94 °C for 5 min and then 30 cycles of 45 s at 94 °C, 45 s at 55 °C and 1 min at 72 °C, followed by 7 min at 72 °C. The PCR products were analyzed on a 1.2% agarose gel and purified from the gel using AxyPrepTM DNA Gel Extraction Kit (Axygen, Suzhou, China), then cloned into pMD 18-T vector (Takara) and sequenced using ABI 3730xl DNA Analyzer (Applied Biosystems, Foster City, USA) repeatedly. The sequence was analysed by the BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) program for homology search, and the predicted amino acid sequence was compared with reported enolase sequences from a wide range of species. Additionally, the conserved motifs, signal peptides, transmembrane regions and glycosyl-phosphatidyl anchor sites of the enolase sequence were analysed by the online resources, including NCBI conserved domains (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi), SignalP 4.1 Server (http://www.cbs.dtu.dk/services/SignalP), TMHMM Server v. 2.0 (http://www.cbs.dtu.dk/services/TMHMM-2.0/), and GPI-SOM (http://gpi.unibe.ch/) [24, 25]. The phylogenetic evolutionary analysis of amino acid sequences of CpEno and enolases of other organisms, such as flagellate protozoa, nematodes, tapeworms, flukes, bacteria, rabbit and human, were determined using MEGA 7.0 software [26].
Expression and purification of recombinant CpEno (rCpEno)
The cpeno gene was digested from recombinant plasmids pMD18T-cpeno by EcoRI and HindIII and inserted into the prokaryotic expression vector pET-28a (+) (Novagen, La Jolla, CA, USA). Recombinant plasmid pET28a-cpeno was transformed into BL21 (DE3) competent cells, and the transformed bacteria were induced using isopropyl-β-D-1-thiogalactopyranoside (IPTG) at a final concentration of 1 mM for 6 h at 37 °C. The rCpEno was purified under native conditions by Ni-NTA His Bind Resin affinity chromatography (Novagen) according to the manufacturer’s instructions. The purified rCpEno was resolved on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and quantified by Pierce™ BCA Protein Assay Kit (Pierce, Rockford, lL, USA).
Western blot analysis
For Western blot analysis, the purified rCpEno was separated on 12% SDS-PAGE and electrotransferred to a PVDF membrane (Merck Millipore, Billerica, MA, USA) using a Trans-Blot® SD Semi-Dry Transfer Cell (Bio-Rad, Hercules, CA, USA) according to standard procedures. Induced pET28a (+) plasmid transformed BL21 (DE3) competent cells were selected as the control. The membranes were blocked with 5% skim milk (Oxoid, Basingstoke, Hants, UK) for 2 h at room temperature and incubated with His-Tag mouse mAb (1:1,000) (Cell Signaling Technology, Danvers, MA, USA) or serum isolated from cattle artificially infected with C. parvum (1:200) for 1 h, followed by incubation with anti-mouse IgG (whole molecule)-Peroxidase conjugate antibody (Sigma-Aldrich, Louis, MO, USA) or anti-bovine IgG (Sigma-Aldrich) at 1:2,000 dilution for 1 h at room temperature. Reaction bands were detected using a DAB Substrate Kit (Pierce).
Polyclonal antibody production
New Zealand rabbits were purchased from Slack Laboratory Animal Co., Ltd., (Shanghai, China). Before immunisation, faeces were collected from the rabbits for 3 days to check for Cryptosporidium spp. infection. One millilitre of purified rCpEno (1 mg/ml) emulsified with the same dose of Freund’s complete adjuvant (Sigma-Aldrich) was injected once into the Cryptosporidium spp. free rabbits, and then 1 ml rCpEno (1 mg/ml) emulsified with Freund’s incomplete adjuvant (Sigma-Aldrich) was injected the next two times at 2-week intervals. Blood samples were collected, and the titers of rabbit antiserum against rCpEno were evaluated using enzyme-linked immunosorbent assay (ELISA). ELISA plate wells were coated with purified rCpEno (2 μg/ml) and incubated overnight at 4 °C. Rabbit muscle enolase (Sigma-Aldrich) was used as a negative control in this experiment. Then the plate wells were blocked with skim milk in PBST (Tween 0.05%) at 37 °C for 1 h. Rabbit anti-C. parvum enolase antibody with different concentrations were added to the wells and finally incubated with horseradish peroxidase (HRP) conjugated anti-rabbit IgG antisera (Sigma-Aldrich). Reactions were developed with tetramethylbenzidine (TMB) substrate solution (Sigma-Aldrich) and terminated with 2 M H2SO4. The values of OD450 were read by an automated ELISA reader (BioTek, Winooski, VT, USA).
Protein immunolocalization in oocysts
Immunolocalization of enolase in C. parvum oocysts was analysed as previously described by Paluszynski et al. [27] with minor modifications. Briefly, 50 μl of the purified oocysts (approximately 105 oocysts/ml) were added to pre-cleaned glass slides and air-dried. Then, the slides were fixed with 4% absolute paraformaldehyde in PBS for 15 min at room temperature. Oocysts were permeabilized with 1% Triton X-100 in 1% bovine serum albumin (BSA) for 15 min and blocked with 1% BSA overnight. The slides were incubated with polyclonal antibodies specific for rCpEno (1:100 dilution in 1% BSA) and washed three times in PBS. Negative controls were treated in the same way but using BSA and pre-immunization antisera. Fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG as secondary antibody (1:1,000 dilution in 1% BSA) (Life Technologies, NY, USA) was added to the slides and incubated for 1 h at 37 °C. After washing three times with PBS, 4’,6’-diamidino-2-phenylindole (DAPI) (Life Technologies) was used to label nuclei for 15 min at 4 °C. The slides were observed by a Nikon Eclipse 80i fluorescence microscope with a DS-Fi1 colour cooled digital camera (Nikon, Tokyo, Japan).
Enzyme activity and kinetic assay
The enzyme activities of rCpEno were determined by measuring the conversion of 2-PGA (Sigma-Aldrich) to PEP (Sigma-Aldrich) at 25 °C, pH 7.4 as described by Han et al. [28] with minor modifications. In brief, 1 mM 2-PGA was added to a 2 ml reaction system, and 20 μg rCpEno was added to the reaction buffer. PEP release values were monitored by NanoDrop 2000C at 240 nm for 40 s intervals in 10 min. As the experiment control, rabbit muscle enolase (positive control) and BSA (negative control) were used in place of rCpEno for the assay.
For the enzyme kinetics research of rCpEno, different concentrations of 2-PGA (0.125, 0.25, 0.5, 1.0, 1.5, 1.75 and 2.0 mM) were added to the reaction buffer. Then, 20 μg rCpEno was added for the assay. The production of PEP was tested at an absorbance value of 240 nm at 1 min intervals for 10 min. The Michaelis constant (K m ) and the maximum reaction velocity (V max ) for rCpEno were determined from double-reciprocal Lineweaver-Burk plots.
Effects of pH, temperature and metal ions on enolase activity
The effects of external factors on the enzyme activity of rCpEno were tested under the following conditions. For the power of hydrogen effects, different pH values (5.5, 6.0, 6.5, 7.0, 7.5, 8.0 and 8.5) were used for the assay at room temperature. To examine the effect of reaction temperature, rCpEno activity was tested at 4 °C, 22 °C, 37 °C and 46 °C at pH 7.4. For the metal ions influence, different concentrations of Ca2+ (0, 20, 40, 80 and 160 mM), K+ (0, 100, 200, 300 and 400 mM), Mg2+ (0, 25, 50, 100 and 200 mM) and Na+ (0, 50, 100, 150, 200, 250 and 300 mM) were measured at 25 °C, pH 7.4. Enolase activities in each of the above tests were compared under the same condition involving 1.0 mM 2-PGA substrate and 20 μg of rCpEno in a 2 ml reaction system. All experiments were repeated three times.
Binding activity of rCpEno to human plasminogen
ELISA plate binding assay was used to determine the binding activity of rCpEno to human plasminogen as described by Bao et al. [16]. Briefly, 10 μg/well of purified rCpEno was added into 96-well plate and incubated at 4 °C overnight. BSA (10 μg/well) was used as a negative control. After being washed three times with PBST, the plate wells were blocked with 5% skim milk (Oxoid) in PBST at 37 °C for 2 h. After washing, different concentrations of human plasminogen (0, 1, 5, 10, 15, 20, 25 and 30 μg/ml in PBST) (Merck Millipore) were added to each well and incubated at 37 °C for 2 h. Then the plates were incubated with rabbit anti-plasminogen polyclonal antibody (100 μl/well, 1: 3,000) (Abcam, MA, USA) at 37 °C for 1.5 h. After washing three times with PBST, the plate wells were incubated with goat anti-rabbit IgG-HRP (100 μl/well, 1: 5,000) (Sigma-Aldrich) at 37 °C for 1 h. Finally, the colour reaction was performed by adding soluble TMB substrate solution and terminated with 2 M H2SO4. Absorbance at OD450 was read using a spectrophotometer (BioTek).
Statistical analysis
Antibody levels and enolase activities were performed using GraphPad Prism 6.0 software for Windows (GraphPad Software, La Jolla, CA, USA), and the data in this study were the mean values ± standard deviations. The significance analysis of differences between 0 mM metal ions and other concentrations at the same time point were analysed by IBM SPSS Statistics V21.0 for Windows (International Business Machines Corp, New York, USA). The differences were considered significant when P < 0.05 by one-way ANOVA test.
Results
Sequence and phylogenetic analysis of CpEno
With cDNA from C. parvum oocysts as the template, a 1,350 bp gene was amplified successfully as theoretically predicted (Additional file 1: Figure S1). BLAST analysis demonstrated that the sequence had 100% identity with the reported C. parvum Iowa II enolase gene sequence (GenBank XM_626138), and encoded a 449 amino acid protein with a theoretical molecular weight of 48.6 kDa. The CpEno amino acid sequence showed 82.1% homology with the C. muris enolase sequence (GenBank XM_002139230). The highest homology of amino acid sequences mainly came from apicomplexan protozoa, such as Eimeria tenella, Theileria annulata, T. gondii, Neospora caninum and P. falciparum, which exhibited 68.0, 67.9, 67.3, 66.9 and 66.8% similarities, respectively (Fig. 1).

Alignment of amino acid sequence of CpEno and other species. The GenBank Accession nos. of these species are as follows: C. parvum (XM_626138), C. muris (XM_002139230), E. tenella (AF353515), T. gondii ENO2 (AY155668), T. annulata (HQ646253), N. caninum (XM_003883950), P. falciparum (U00152), A. thaliana (AF424603) and Human gamma-2 enolase (M22349). Similar to A. thaliana, a plant-like five-amino-acid sequence was present in apicomplexan parasites (shown in the red box) and was not found in the human sequence. The Mg2+ binding motifs are shown in a blue box, substrate-binding motifs are shown in a green box, plasminogen binding motifs are shown in a black box, and the enolase signature is underlined in red
CpEno also had a plant-like five-amino-acid sequence (EYGFT) between sites 108 to 112, which was similar to others apicomplexan parasites (Fig. 1). Using online server analysis, CpEno showed a high degree of conservation with other species, including full conservation of the Mg2+ binding motifs (S41, D255, E304 and D331), substrate-binding motifs (H168, E220, K356, 384HRS386 and K407) and the enolase signature (353LLLKVNQIGSVTEA366) (Fig. 1). Similar to the Streptococcus pneumonia enolase amino acid sequence (248FYDKERKVY256) [14], a plasminogen binding motif (261FFVEDKKCY269) was also found in CpEno and other species (Fig 1).
Phylogenetic analysis showed that the C. parvum enolase sequence was closest to C. muris, and then other apicomplexan protozoa. All of them located in the same branch. In all selected animals, CpEno had a closer relationship with nematodes, tapeworms, flukes and mammals (human and rabbit) than with flagellate protozoa and bacteria (Fig. 2).

Phylogenetic analysis of CpEno with other enolase sequences. The phylogenetic tree was constructed using the neighbor-joining method by MEGA 7 software. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) are shown next to the branches. The analysis involved 23 amino acid sequences, and all the apicomplexan protozoa located in the same branch (red box)
Expression and Western blot analysis
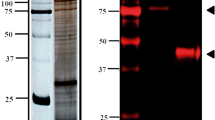
Using T4 DNA ligase (TaKaRa), cpeno was ligated into the pET-28a (+) vector and the recombinant plasmid was transformed into competent E. coli BL21 (DE3) to express the recombinant fusion protein after inducing with IPTG. The protein was analysed by SDS-PAGE and purified by Ni-NTA His Bind Resin affinity. Coomassie staining of the gels showed that most rCpEno remained within the supernatant (Additional file 2: Figure S2, Lanes 3 and 4), as well as a single band of 52.5 kDa protein, in agreement with the expected molecular mass including the 6 × His-tagged recombinant polypeptide (Additional file 2: Figure S2, Lanes 5 and 6). The recombinant protein was further characterised by Western blot detection with His-Tag mouse mAb and positive serum isolated from Cryptosporidium infected cattle (1:200) (Additional file 3: Figure S3). The results confirmed the recombinant protein could react with the two antibodies tested. In contrast, the induced pET28a (+) plasmid transformed E. coli BL21 (DE3) did not react with the positive serum or His-Tag mouse mAb.
Anti-rCpEno polyclonal antibody preparation
Purified rCpEno was used to generate rabbit polyclonal antisera. One week after the third immunisation, the reactivity and specificity of antibodies were tested by ELISA and western blot. The antibody titer of immune serum was more than 1/102,400. At that dilution, the optical density (OD) of immune sera was also higher than 2.0. While using rabbit muscle enolase as a negative control, no reaction was found when the dilution of immune serum was more than 1:6400 (Fig. 3a). A single band of about 52.5 kDa was recognised by immune serum using Western blot assay, and the induced pET28a (+) plasmid transformed E. coli BL21 (DE3) did not react with the immune serum (Fig. 3b).

Polyclonal antibody detection of rCpEno immune serum. Rabbit polyclonal antisera were collected after the third immunisation. The reactivity and specificity of antibodies were tested by ELISA (a) and Western blot (b). A series of diluted antibodies were examined for their specific binding activity to either rCpEno or rabbit muscle enolase (negative control). A good antibody titer was detected when the dilution multiple was higher than 1/102,400, and a 52.5 kDa specific band was observed by western blot assay. Data shown are the mean values ± standard deviations
The localisation of CpEno in C. parvum oocyst
Freshly collected oocysts of C. parvum were fixed and treated with rabbit anti-rCpEno sera. The detected proteins were visualised by fluorescence microscopy using FITC-labelled secondary antisera against rabbit immunoglobulins. Clear signals were found surrounding the oocysts incubated with rabbit anti-rCpEno serum indicating that the protein localised on the surface (Fig. 4a). No signals were obtained after treatment of the slide with BSA (Fig. 4b) or pre-immunization antisera (data not shown).

Immunolocalization of enolase in C. parvum oocysts. Oocysts were incubated with rabbit anti-enolase serum (a) and a BSA (negative control) (b). Enolases were located on the surface of oocysts (a), and no signals were found with BSA (b). Different fluorescent probes were used, rabbit anti-CpEno (green) and DAPI (nuclear marker) (blue). Arrows indicate C. parvum oocysts. Scale-bars: 5 μm
Enzyme activity and effects of pH, temperature and metal ions
The enzyme activity of rCpEno was detected by NanoDrop 2000C at 240 nm by increasing the amount of 2-PGA to PEP. The activity of rCpEno in catalysing the conversion of 2-PGA to PEP was lower than rabbit muscle enolase; however, almost no reaction was detected in BSA (Fig. 5a). Kinetic measurements using 2- PGA as the substrate gave a specific activity of 33.5 U/mg. A Michaelis-Menten plot was fitted to the equation V = V max S/(S + K m ), and the K m for the forward reaction was 0.571 mM/l, the V max was 0.054 mM/l · min (Fig. 5b).

Enzymatic characterization of rCpEno. a Enzymatic activity of rCpEno was determined by measuring the conversion of 2-PGA to PEP. The absorbance value of rCpEno at 240 nm was lower than rabbit muscle enolase (positive control), and nearly no value was detected in BSA (negative control) at the same time point. b Enzyme kinetic research of rCpEno. The conversion rate (V) of 2-PGA (S) to PEP was measured at 240 nm using 20 μg of rCpEno and various concentrations of 2-PGA (0.125–2 mM). K m and V max for rCpEno were determined as 0.571 mM/l and 0.054 mM/l · min, respectively, using a Lineweaver-Burk double-reciprocal plot
With regards to external influencing factors, pH 7.0 and pH 7.5 were found to be the most suitable pH value (Fig. 6a). Minor effects on the activity of rCpEno were found when the temperature changed from 4 °C to 46 °C (ANOVA: F (3,8) = 3.273, P = 0.065) (Fig. 6b). The rCpEno activity was inhibited with increase of Ca2+(ANOVA: F (5,72) = 18.719, P < 0.0001), K+(ANOVA: F (4,60) = 21.980, P < 0.0001), Mg2+(ANOVA: F (3,48) = 9.580, P < 0.0001) and Na+(ANOVA: F (5,72) = 11.947, P < 0.0001) concentrations (Fig. 7), while low concentrations of Mg2+ (25 mM) had no obvious effect on enzyme activity (ANOVA: F (1,24) = 0.163, P = 0.690) (Fig. 7c).

Effect of pH and temperature on the catalytic/enzymatic activity of rCpEno. Various pH (5.5–8.5) and temperature (4 °C, 22 °C, 37 °C and 46 °C) were determined to measure enzymatic activities using 20 μg of rCpEno at 240 nm. The maximal enzymatic reactivity of rCpEno occurred at pH 7.0 to 7.5 (a), and minor effects were found at temperatures from 4 °C to 46 °C (b)

Effect of metal ions on the catalytic/enzymatic activity of rCpEno. Different concentrations of metal ions (Ca2+, K+, Mg2+ and Na+) (a, b, c and d) were measured at room temperature and pH 7.4. The reaction mixtures consisted of 20 μg purified rCpEno and one mM 2-PGA. The enzymatic activities of rCpEno were inhibited with increased concentrations of metal ions (P < 0.05). However, low concentrations of Mg2+ had almost no influence on enzyme activity (P > 0.05). The experiments were performed in triplicate, and the data shown are the mean values ± standard deviations
Binding assay of rCpEno to human plasminogen
An ELISA experiment confirmed that rCpEno could bind to human plasminogen in a dose-dependent pattern. BSA showed no interaction with human plasminogen (Fig. 8).

Binding activity of rCpEno to human plasminogen. The binding ability of rCpEno to human plasminogen was tested by ELISA. CpEno could bind to human plasminogen, and the binding activities of rCpEno were raised along with the concentrations of plasminogen increasing. BSA was not able to bind to human plasminogen
Discussion
Cryptosporidium parvum is one of the most common species in the genus to infect mammalian intestinal epithelial cells (IECs) with the onset of diarrhoea as a remarkable characteristic [29]. Genome sequencing and biochemical data have revealed that C. parvum lacks many mitochondrial proteins, including those required for the TCA cycle, fatty acid oxidation [8]. However, some essential metabolic pathways are still present in C. parvum. Glycolysis as the source of energy and carbon seems to play an important role in these pathways [30]. In addition to being an essential glycolytic enzyme, several studies have shown that enolase is a multifunctional protein, which participates in many important processes, such as motility, adhesion, invasion, differentiation, development and gene regulation in protozoa, including E. tenella [31, 32], Entamoeba histolytica [33], Giardia lamblia [34], Leishmania donovani [35], Plasmodium spp. [21, 36,37,38,39,40,41,42,43,44,45,46,47,48], T. gondii [20, 49, 50], Trypanosoma spp. [51] and Trichomonas vaginalis [52]. However, to our knowledge, the location and characteristics of C. parvum enolase are still unclear.
The complete encoding gene of CpEno was amplified from C. parvum cDNA and sequenced in this study. Sequence analysis showed that CpEno had 82.1% identity with C. muris enolase, a high degree of conservation with apicomplexan parasites (from 66.8 to 68.0% sequence identity), and 54.7 to 65.2% with other species. CpEno also has many conserved sites, such as metal- and substrate-binding motifs and an enolase signature which is similar to that in Schistosoma bovis and Ornithodoros moubata [53, 54], indicating that this protein is conserved in different phylum in the animal kingdom.
Sequence and phylogenetic analysis also showed that CpEno and other apicomplexan protozoa enolases were located in the same branch, and shared a plant-like pentapeptide insert (i.e. C. parvum 106EYGFT110, C. muris 103EYGYN107, E. tenella 103EWGWS107, N. caninum 103EWGYS107, P. falciparum 104EWGWS108, T. annulata 103EWGYC107, T. gondii ENO1 103EWGYS107 and T. gondii ENO2 103EWGWC107) [31, 55,56,57,58,59], which is more closely related to plant enolase (i.e. A. thaliana 103EWGYC107) than to the mammalian counterpart. Many studies have reported that this site is important for activities of enolases in apicomplexan protozoa. Vora et al. [39] found that deletion of this insert resulted in a 100-fold decrease of full enzyme activity and caused dissociation of the dimeric form into monomers in P. falciparum. A similar deletion provoked a decrease of substrate affinity in T. gondii ENO1 [57]. Recent studies showed that this motif could stabilise P. falciparum enolase in an active form and was also a protective antigenic epitope [46, 47]. Mukherjee et al. reported that a deletion variant Δ-rPfeno (P. falciparum enolase lacking insert sequence 104EWGWS108) was incapable of binding to PWWP (a conserved motif of the ookinete sequence) peptides, implying that this insert was involved in mosquito midgut invasion [48]. Because this unique region is absent in human enolase, it may be a potential drug target site for inhibitor design. However, the function of this insert in CpEno needs further studies.
According to the C. parvum genome sequence (http://cryptodb.org/cryptodb/), we found that CpEno was a single gene, which was similar to most studies of apicomplexan parasites, such as E. tenella [31], Plasmodium spp. [37, 43, 55], T. annulata [59], and was in contrast to studies of T. gondii enolase which reported two stage-specific glycolytic enolases, ENO1 and ENO2 [20, 56, 57].
In this study, the purified rCpEno could convert 2-PGA to PEP and the reaction activity was 33.5 U/mg, which was similar with P. falciparum (30 ± 3 U/mg) and T. annulata (40 U/mg) [59, 60]. Compared with other apicomplexan parasites, the K m value of rCpEno (0.571 mM) was higher than in P. falciparum (0.041 ± 0.004 mM), T. gondii ENO1 (0.0768 ± 0.0064 mM), T. gondii ENO2 (0.0777 ± 0.0049 mM) and T. annulata (0.106 mM) [57, 59, 60].
With regards to environmental effect factors, rCpEno showed a stable temperature characteristic. Minor effects were observed on enzyme activity at different temperatures which were similar to some other species, such as Brucella abortus and Schistosoma japonicum [28, 61]. In addition, the most suitable pH value for rCpEno was 7.0–7.5, this result was consistent with previous studies showing that maximal enolase activity was observed at pH 7.4–7.6 in P. falciparum and pH 7.2 ± 0.1 in T. gondii [57, 60]. Compared with 0 mM salt ions (Ca2+, K+, Mg2+ and Na+), the enzyme activity was inhibited by increasing concentrations of metal ions (P < 0.05) while there was no obvious difference at 25 mM Mg2+ compared with 0 mM Mg2+ at the same time point (P > 0.05), which implied that low concentrations of Mg2+ (25 mM) had almost no effect on enzyme activity. This result was different with a study in P. falciparum by Pal-Bhowmick et al. [60] who reported the activity of P. falciparum enolase was inhibited by Na+, whereas K+ had a slight activating effect. They also found that low concentrations of Mg2+ had apparent activation effect and high concentrations had an inhibitory effect [60]. Previous research showed that Ca2+ was a “nonactivating” metal ion which inhibited the enzyme activity by replacing the catalytic Mg2+ ions [62]. In yeast enolase, two binding sites of Mg2+ have been reported, site I is a conformational site that could induce structural changes, and site II is a catalytic site that could promote catalytic activity [62, 63]. In Trypanosoma brucei enolase, another Mg2+ binding site had been reported, which was a low-affinity site involved in inhibition [64]. However, the function of Mg2+ binding sites in CpEno are still unclear, and there is a need for further crystal structure studies of CpEno. To our knowledge, this is the first report of the catalytic activity, reaction constant and effect factors for C. parvum enolase.
High titers of specific antibodies were induced after three immunisations with rCpEno, and good reactogenicity and immunogenicity were seen by ELISA and western blot. Using the antisera as a primary antibody, surface localisation of enolase on oocysts was revealed by fluorescence microscopy, which was consistent with previous studies in Plasmodium spp. where enolase located on the cell surface [36,37,38, 43]. However, in E. tenalla and T. gondii, enolases were also found in the nucleus [31, 49]. Due to the low number of fresh oocysts acquired for this research, the specific localisation of CpEno on sporozoites is still unclear. Further experiments on this aspect need to be further performed in the future.
Given the paucity of therapeutic drugs and the lack of effective vaccines, it is important to identify and characterise the specific surface molecules mediating attachment and invasion of the parasite into host cells [30]. Membrane proteins are involved in various critical biological processes [65], which are believed to mediate attachment and invasion of Cryptosporidium. In the present study, our first identification of CpEno on the surface by immunofluorescence examinations indicates that this protein may be a membrane-related protein. This was also confirmed in our laboratory when identifying membrane proteins of C. parvum sporozoites (unpublished). Therefore, along with a role as an essential glycolytic enzyme, CpEno may be involved in the attachment and/or invasion of IECs as in other organisms, including some apicomplexan protozoa. Previous studies have reported that enolase is one of the most frequent surface-localized glycolytic enzymes in many species, such as Borrelia burgdorferi, G. duodenalis, Leishmania mexicana, Mycoplasma bovis, M. synoviae S. bovis, S. japonicum, Streptococcus pneumoniae, T. vaginalis and Vibrio parahaemolyticus [12,13,14,15,16,17, 34, 52, 53, 61, 66]. In these pathogens, surface enolase plays an important role in host-pathogen interactions by binding plasminogen and is also possibly involved in pathogen invasion. In this study, a plasminogen binding assay showed that rCpEno could bind to human plasminogen, in agreement with studies on other species [12,13,14,15,16,17, 52, 53]. This suggests that rCpEno may participate as a parasitic virulence factor for Cryptosporidium parvum.
The publication of the C. parvum genome has enhanced our understanding on the parasite, but the function of a specific gene study was hindered for a long period in the past by the lack of a C. parvum long-term culture in vitro and a missing toolbox for genetic manipulation [67]. It is encouraging that the first system for genetic modification of Cryptosporidium was described, and a stable C. parvum transgenic line was established through a clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 system [68]. Using this system, research into the functions of enolase in C. parvum will be achieved through genetic manipulation and will help us deeply understand the role of enolase in the relationship between the parasite and host.
Conclusions
In summary, a 1,350 bp enolase encoding gene was amplified from C. parvum cDNA and expressed in E. coli BL21 (DE3) to produce a recombinant protein rCpEno with a molecular weight of 52.5 kDa. Enzyme activity analysis showed that the reaction activity of rCpEno of converting 2-PGA to PEP was 33.5 U/mg and the most suitable pH value was 7.0–7.5. The K m and V max were 0.571 mM/l and 0.054 mM/l · min, respectively. The activities were inhibited with an increased concentration of metal ions, but minor effects were detected with temperature changes. Immunofluorescence assay showed that rCpEno localised on the surface of oocysts. The binding assay demonstrated that rCpEno could bind to human plasminogen with a dose-dependent pattern. To our knowledge, this is the first report describing the characteristics, binding activity and localisation of C. parvum enolase. The surface localisation of C. parvum enolase implied that it is not only a glycolytic enzyme but also participates in other processes of the parasite.
Abbreviations
- 2-PGA:
-
2-phosphoglycerate
- BSA:
-
Bovine serum albumin
- CpEno:
-
C. parvum enolase
- cpeno :
-
C. parvum enolase gene
- CRISPR:
-
Clustered regularly interspaced short palindromic repeat
- DAB:
-
3,3’-diaminobenzidine
- DAPI:
-
4’,6’-diamidino-2-phenylindole
- ELISA:
-
Enzyme-linked immunosorbent assay
- FITC:
-
Fluorescein isothiocyanate
- HRP:
-
Horseradish peroxidase
- IECs:
-
Intestinal epithelial cells
- IPTG:
-
Isopropyl-β-D-1-thiogalactopyranoside
- K m :
-
Michaelis constant
- OD:
-
Optical density
- PBS:
-
Phosphate buffer saline
- PBST:
-
Phosphate buffer saline (Tween 0.05%)
- PCR:
-
Polymerase chain reaction
- PEP:
-
Phosphoenolpyruvate
- rCpEno:
-
Recombinant C. parvum enolase
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- SDS-PAGE:
-
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- TCA:
-
Tricarboxylic acid
- TMB:
-
Tetramethylbenzidine
- V max :
-
Maximum reaction velocity
References
Xiao L. Molecular epidemiology of cryptosporidiosis: an update. Exp Parasitol. 2010;124:80–9.
Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case–control study. Lancet. 2013;382:209–22.
Striepen B. Parasitic infections: Time to tackle cryptosporidiosis. Nature. 2013;503:189–91.
Matsubayashi M, Teramoto-Kimata I, Uni S, Lillehoj HS, Matsuda H, Furuya M, et al. Elongation factor-1α is a novel protein associated with host cell invasion and a potential protective antigen of Cryptosporidium parvum. J Biol Chem. 2013;288:34111–20.
Bhalchandra S, Ludington J, Coppens I, Ward HD. Identification and characterization of Cryptosporidium parvum Clec, a novel C-type lectin domain-containing mucin-like glycoprotein. Infect Immun. 2013;81:3356–65.
Hannaert V, Bringaud F, Opperdoes FR, Michels PA. Evolution of energy metabolism and its compartmentation in Kinetoplastida. Kinetoplastid Biol Dis. 2003;2:11.
Rider Jr SD, Zhu G. Cryptosporidium: genomic and biochemical features. Exp Parasitol. 2010;124:2–9.
Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, et al. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science. 2004;304:441–5.
Thompson RC, Olson ME, Zhu G, Enomoto S, Abrahamsen MS, Hijjawi NS. Cryptosporidium and cryptosporidiosis. Adv Parasitol. 2005;59:77–158.
Pancholi V. Multifunctional α-enolase: its role in diseases. Cell Mol Life Sci. 2001;58:902–20.
Liu KJ, Shih NY. The role of enolase in tissue invasion and metastasis of pathogens and tumor cells. J Cancer Mol. 2007;3:45–8.
Nogueira SV, Smith AA, Qin JH, Pal U. A surface enolase participates in Borrelia burgdorferi-plasminogen interaction and contributes to pathogen survival within feeding ticks. Infect Immun. 2012;80:82–90.
Song Z, Li Y, Liu Y, Xin J, Zou X, Sun W. α-enolase, an adhesion-related factor of Mycoplasma bovis. PLoS ONE. 2012;7:e38836.
Bergmann S, Wild D, Diekmann O, Frank R, Bracht D, Chhatwal G, et al. Identification of a novel plasmin(ogen)-binding motif in surface displayed α-enolase of Streptococcus pneumoniae. Mol Microbiol. 2003;49:411–23.
Bergmann S, Schoenen H, Hammerschmidt S. The interaction between bacterial enolase and plasminogen promotes adherence of Streptococcus pneumoniae to epithelial and endothelial cells. Int J Med Microbiol. 2013;303:452–62.
Bao S, Guo X, Yu S, Ding J, Tan L, Zhang F, et al. Mycoplasma synoviae enolase is a plasminogen/fibronectin binding protein. BMC Vet Res. 2014;10:223.
Jiang W, Han X, Wang Q, Li X, Yi L, Liu Y, et al. Vibrio parahaemolyticus enolase is an adhesion-related factor that binds plasminogen and functions as a protective antigen. Appl Microbiol Biotechnol. 2014;98:4937–48.
Morita T, Kawamoto H, Mizota T, Inada T, Aiba H. Enolase in the RNA degradosome plays a crucial role in the rapid decay of glucose transporter mRNA in the response to phosphosugar stress in Escherichia coli. Mol Microbiol. 2004;54:1063–75.
Lee H, Guo Y, Ohta M, Xiong L, Stevenson B, Zhu JK. LOS2, a genetic locus required for cold-responsive gene transcription encodes a bi-functional enolase. EMBO J. 2002;21:2692–702.
Mouveaux T, Oria G, Werkmeister E, Slomianny C, Fox BA, Bzik DJ, et al. Nuclear glycolytic enzyme enolase of Toxoplasma gondii functions as a transcriptional regulator. PLoS ONE. 2014;9:e105820.
Alves E, Maluf FV, Bueno VB, Guido RV, Oliva G, Singh M, et al. Biliverdin targets enolase and eukaryotic initiation factor 2 (eIF2α) to reduce the growth of intraerythrocytic development of the malaria parasite Plasmodium falciparum. Sci Rep. 2016;6:22093.
Chen Z, Mi R, Yu H, Shi Y, Huang Y, Chen Y, et al. Prevalence of Cryptosporidium spp. in pigs in Shanghai, China. Vet Parasitol. 2011;181:113–9.
Arrowood MJ, Sterling CR. Isolation of Cryptosporidium oocysts and sporozoites using discontinuous sucrose and isopycnic Percoll gradients. J Parasitol. 1987;73:314–9.
Marchler-Bauer A, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, et al. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009;37:D205–10.
Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–6.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Paluszynski J, Monahan Z, Williams M, Lai O, Morris C, Burns P, et al. Biochemical and functional characterization of CpMuc4, a Cryptosporidium surface antigen that binds to host epithelial cells. Mol Biochem Parasitol. 2014;193:114–21.
Han X, Ding C, Chen H, Hu Q, Yu S. Enzymatic and biological characteristics of enolase in Brucella abortus A19. Mol Biol Rep. 2012;39:2705–11.
McDonald V, Korbel DS, Barakat FM, Choudhry N, Petry F. Innate immune responses against Cryptosporidium parvum infection. Parasite Immunol. 2013;35:55–64.
Borad A, Ward H. Human immune responses in cryptosporidiosis. Future Microbiol. 2010;5:507–19.
Labbé M, Péroval M, Bourdieu C, Girard-Misguich F, Péry P. Eimeria tenella enolase and pyruvate kinase: a likely role in glycolysis and in others functions. Int J Parasitol. 2006;36:1443–52.
Liu L, Chen Z, Mi R, Zhang K, Liu Y, Jiang W, et al. Effect of Acetamizuril on enolase in second-generation merozoites of Eimeria tenella. Vet Parasitol. 2016;215:88–91.
Tovy A, Siman Tov R, Gaentzsch R, Helm M, Ankri S. A new nuclear function of the Entamoeba histolytica glycolytic enzyme enolase: the metabolic regulation of cytosine-5 methyltransferase 2 (Dnmt2) activity. PLoS Pathog. 2010;6:e1000775.
Castillo-Romero A, Davids BJ, Lauwaet T, Gillin FD. Importance of enolase in Giardia lamblia differentiation. Mol Biochem Parasitol. 2012;184:122–5.
Swenerton RK, Zhang S, Sajid M, Medzihradszky KF, Craik CS, Kelly BL, et al. The oligopeptidase B of Leishmania regulates parasite enolase and immune evasion. J Biol Chem. 2011;286:429–40.
Pal-Bhowmick I, Mehta M, Coppens I, Sharma S, Jarori GK. Protective properties and surface localization of Plasmodium falciparum enolase. Infect Immun. 2007;75:5500–8.
Pal-Bhowmick I, Vora HK, Jarori GK. Sub-cellular localization and post-translational modifications of the Plasmodium yoelii enolase suggest moonlighting functions. Malar J. 2007;6:45.
Bhowmick IP, Kumar N, Sharma S, Coppens I, Jarori GK. Plasmodium falciparum enolase: stage-specific expression and sub-cellular localization. Malar J. 2009;8:179.
Vora HK, Shaik FR, Pal-Bhowmick I, Mout R, Jarori GK. Effect of deletion of a plant like pentapeptide insert on kinetic, structural and immunological properties of enolase from Plasmodium falciparum. Arch Biochem Biophys. 2009;485:128–38.
Das S, Shevade S, LaCount DJ, Jarori GK. Plasmodium falciparum enolase complements yeast enolase functions and associates with the parasite food vacuole. Mol Biochem Parasitol. 2011;179:8–17.
Ghosh AK, Coppens I, Gårdsvoll H, Ploug M, Jacobs-Lorena M. Plasmodium ookinetes coopt mammalian plasminogen to invade the mosquito midgut. Proc Natl Acad Sci U S A. 2011;108:17153–8.
Ghosh AK, Jacobs-Lorena M. Surface-expressed enolases of Plasmodium and other pathogens. Mem Inst Oswaldo Cruz. 2011;106 Suppl 1:85–90.
Hernández-Romano J, Rodríguez MH, Pando V, Torres-Monzón JA, Alvarado-Delgado A, Lecona Valera A, et al. Conserved peptide sequences bind to actin and enolase on the surface of Plasmodium berghei ookinetes. Parasitology. 2011;138:1341–53.
Shevade S, Jindal N, Dutta S, Jarori GK. Food vacuole associated enolase in Plasmodium undergoes multiple post-translational modifications: evidence for atypical ubiquitination. PLoS ONE. 2013;8:e72687.
Vega-Rodríguez J, Ghosh AK, Kanzok SM, Dinglasan RR, Wang S, Bongio NJ, et al. Multiple pathways for Plasmodium ookinete invasion of the mosquito midgut. Proc Natl Acad Sci U S A. 2014;111:E492–500.
Dutta S, DasSarma P, DasSarma S, Jarori GK. Immunogenicity and protective potential of a Plasmodium spp. enolase peptide displayed on archaeal gas vesicle nanoparticles. Malar J. 2015;14:406.
Dutta S, Mukherjee D, Jarori GK. Replacement of Ser108 in Plasmodium falciparum enolase results in weak Mg(II) binding: role of a parasite-specific pentapeptide insert in stabilizing the active conformation of the enzyme. FEBS J. 2015;282:2296–308.
Mukherjee D, Mishra P, Joshi M, Thakur PK, Hosur RV, Jarori GK. EWGWS insert in Plasmodium falciparum ookinete surface enolase is involved in binding of PWWP containing peptides: implications to mosquito midgut invasion by the parasite. Insect Biochem Mol Biol. 2016;68:13–22.
Ferguson DJ, Parmley SF, Tomavo S. Evidence for nuclear localisation of two stage-specific isoenzymes of enolase in Toxoplasma gondii correlates with active parasite replication. Int J Parasitol. 2002;32:1399–410.
Holmes M, Liwak U, Pricop I, Wang X, Tomavo S, Ananvoranich S. Silencing of tachyzoite enolase 2 alters nuclear targeting of bradyzoite enolase 1 in Toxoplasma gondii. Microbes Infect. 2010;12:19–27.
Avilán L, Gualdrón-López M, Quiñones W, González-González L, Hannaert V, Michels PA, et al. Enolase: a key player in the metabolism and a probable virulence factor of trypanosomatid parasites-perspectives for its use as a therapeutic target. Enzyme Res. 2011;2011:932549.
Mundodi V, Kucknoor AS, Alderete JF. Immunogenic and plasminogen-binding surface-associated α-enolase of Trichomonas vaginalis. Infect Immun. 2008;76:523–31.
de la Torre-Escudero E, Manzano-Román R, Pérez-Sánchez R, Siles-Lucas M, Oleaga A. Cloning and characterization of a plasminogen-binding surface-associated enolase from Schistosoma bovis. Vet Parasitol. 2010;173:76–84.
Díaz-Martín V, Manzano-Román R, Oleaga A, Encinas-Grandes A, Pérez-Sánchez R. Cloning and characterization of a plasminogen-binding enolase from the saliva of the argasid tick Ornithodoros moubata. Vet Parasitol. 2013;191:301–14.
Read M, Hicks KE, Sims PF, Hyde JE. Molecular characterisation of the enolase gene from the human malaria parasite Plasmodium falciparum. Evidence for ancestry within a photosynthetic lineage. Eur J Biochem. 1994;220:513–20.
Dzierszinski F, Popescu O, Toursel C, Slomianny C, Yahiaoui B, Tomavo S. The protozoan parasite Toxoplasma gondii expresses two functional plant-like glycolytic enzymes. Implications for evolutionary origin of apicomplexans. J Biol Chem. 1999;274:24888–95.
Dzierszinski F, Mortuaire M, Dendouga N, Popescu O, Tomavo S. Differential expression of two plant-like enolases with distinct enzymatic and antigenic properties during stage conversion of the protozoan parasite Toxoplasma gondii. J Mol Biol. 2001;309:1017–27.
Pal-Bhowmick I, Vora HK, Roy J, Sharma S, Jarori GK. Generation and characterisation of monoclonal antibodies specific to Plasmodium falciparum enolase. J Vector Borne Dis. 2006;43:43–52.
Cayir E, Erdemir A, Ozkan E, Topuzogullari M, Bolat ZB, Akat A, et al. Cloning of intron-removed enolase gene and expression, purification, kinetic characterization of the enzyme from Theileria annulata. Mol Biotechnol. 2014;56:689–96.
Pal-Bhowmick I, Sadagopan K, Vora HK, Sehgal A, Sharma S, Jarori GK. Cloning, over-expression, purification and characterization of Plasmodium falciparum enolase. Eur J Biochem. 2004;271:4845–54.
Yang J, Qiu C, Xia Y, Yao L, Fu Z, Yuan C, et al. Molecular cloning and functional characterization of Schistosoma japonicum enolase which is highly expressed at the schistosomulum stage. Parasitol Res. 2010;107:667–77.
Lebioda L, Stec B, Brewer JM, Tykarska E. Inhibition of enolase: the crystal structures of enolase-Ca2+-2-phosphoglycerate and enolase-Zn2+-phosphoglycolate complexes at 2.2-Å resolution. Biochemistry. 1991;30:2823–7.
Wedekind JE, Poyner RR, Reed GH, Rayment I. Chelation of serine 39 to Mg2+ latches a gate at the active site of enolase: structure of the bis(Mg2+) complex of yeast enolase and the intermediate analog phosphonoacetohydroxamate at 2.1-Å resolution. Biochemistry. 1994;33:9333–42.
da Silva Giotto MT, Hannaert V, Vertommen D, de A S Navarro MV, Rider MH, Michels PA, et al. The crystal structure of Trypanosoma brucei enolase: visualisation of the inhibitory metal binding site III and potential as target for selective, irreversible inhibition. J Mol Biol. 2003;331:653–65.
Wang L, Tonggu L. Membrane protein reconstitution for functional and structural studies. Sci China Life Sci. 2015;58:66–74.
Quiñones W, Peña P, Domingo-Sananes M, Cáceres A, Michels PA, Avilan L, et al. Leishmania mexicana: molecular cloning and characterization of enolase. Exp Parasitol. 2007;116:241–51.
Lendner M, Daugschies A. Cryptosporidium infections: molecular advances. Parasitology. 2014;141:1511–32.
Vinayak S, Pawlowic MC, Sateriale A, Brooks CF, Studstill CJ, Bar-Peled Y, et al. Genetic modification of the diarrhoeal pathogen Cryptosporidium parvum. Nature. 2015;523:477–80.
Acknowledgements
The authors are grateful to Dr Zhenquan Jia from Department of Biology, The University of North Carolina at Greensboro for his revision the draft of this paper.
Funding
This study was supported in part by Shanghai Agriculture Applied Technology Development Program, China (Grant No. 20150110), National Risk Assessment Project for Quality and Safety of Agricultural Products (Grant No. GJFP201700703), Opening Foundation of State Key Laboratory of Veterinary Etiological Biology (Grant No. SKLVEB2013KFKT017), Basic Foundation for Scientific Research of State-level Public Welfare Institutes of China (Grant No.2016JB13 and 2013JB13), and Foundation of the Agricultural Science and Technology Innovation Program of Chinese Academy of Agricultural Sciences (Grant No. SHVRI-ASTIP-2016-9).
Availability of data and materials
The datasets supporting the conclusions in this article are included within the article.
Authors’ contributions
ZC designed the study and revised the manuscript. RM and XY performed the whole experiments and drafted the manuscript. YH and KL participated the animal immunisation and parasites collection. LC and XH participated protein immunolocalization. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
Animal experiments were approved by the Animal Ethics Committee of Shanghai Veterinary Research Institute. During the whole experimental process, all used protocols were consistent with the rules for Animal Care of the Chinese Academy of Agricultural Sciences. The approval number of Animal Ethics Committee were shvri-co-0002 (cattle) and shvri-ro-0888 (rabbit).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Figure S1.
Amplification of cpeno by RT-PCR. Lane 1: PCR product of cpeno gene amplified from C. parvum cDNA; Lane M: DL2,000 DNA Marker (Takara); Lane N: negative control. (TIF 325 kb)
Additional file 2: Figure S2.
SDS-PAGE of rCpEno. Lane 1: uninduced pET28a-cpeno transfected E. coli BL21; Lane 2: induced pET28a-cpeno transfected E. coli BL21; Lanes 3 and 4: soluble and insoluble fractions of the induced pET28a-cpeno transfected E. coli BL21 extract, respectively; Lanes 5 and 6: purified rCpEno; Lane M: prestained protein ladder (Thermo Fisher Scientific). (TIF 670 kb)
Additional file 3: Figure S3.
Western blot analysis of purified rCpEno. Purified rCpEno and induced pET-28a (+) plasmid transfected E. coli BL21 (DE3) competent cells were incubated with His-Tag mouse mAb (a) and C. parvum positive serum from cattle (b), respectively. Lane M: prestained protein ladder (Thermo Fisher Scientific). (TIF 902 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mi, R., Yang, X., Huang, Y. et al. Immunolocation and enzyme activity analysis of Cryptosporidium parvum enolase. Parasites Vectors 10, 273 (2017). https://doi.org/10.1186/s13071-017-2200-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-017-2200-y