
Abstract
Background
Although effective results of many studies support the use of spinal cord stimulation in chronic pain patients, no randomized controlled trial has been undertaken in China to date. CITRIP is a multicenter, prospective, randomized, withdrawal study designed to evaluate the clinical effectiveness and safety of spinal cord stimulation plus remote programming management in patients with intractable trunk or limb pain.
Method
Participants will be recruited in approximately 10 centers across China. Eligible participants with intractable trunk or limb and an average visual analog scale (VAS) score ≥ 5 will undergo a spinal cord stimulation test. Participants with VAS score reduction ≥ 50% could move forward to receive implantation of an implanted pulse generator. In the withdrawal period at 3-month follow-up visit, participants randomized to the experimental group (EG) will undergo continuous stimulation while ceasing the stimulation in the control group (CG). The outcome assessment will occur at baseline and at 1, 3 (pre- and post-randomization), and 6 months. The primary outcome is the difference of maximal VAS score between EG and CG in the withdrawal period compared with baseline before the withdrawal period. Additional outcomes include VAS score change at 1-, 3-, and 6-month follow-ups; responder rate (VAS score improving by 50%); achievement rate of a desirable pain state (VAS score ≤ 4); awake times during sleep; Beck Depression Inventory for depression evaluation; short-form 36 for quality of life evaluation; drug usage; and satisfaction rating of the device. Adverse events will be collected. The primary analysis will follow the intention-to-treat principle.
Discussion
The CITRIP study seeks to evaluate the effectiveness and safety of a randomized withdrawal trial of spinal cord stimulation for patients with intractable trunk or limb pain.
Trial registration
ClinicalTrials.gov NCT03858790. Registered on March 1, 2019, retrospectively registered
Similar content being viewed by others


Background
Chronic pain is a major clinical, social, and economic problem that appears to have a great negative influence on the quality of life. In 2010, chronic pain costs the USA approximately $635 billion in health care, which exceeds the total amount of cancer and cardiovascular and cerebral vascular diseases [1].
Chronic intractable pain (CIP) refers to the pain lasting for over 3 months and is refractory to conservative treatments, including oral medications, nerve block, epidural corticosteroids, physical and psychological rehabilitation therapy, and chiropractic care [2]. Since 1967, spinal cord stimulation (SCS) was used to manage chronic intractable pain, such as failed back surgery syndrome (FBSS) [3, 4], complex regional pain syndrome [5], and ischemic vascular disease-related pain [6]. Some systematic reviews also suggested that SCS has strong evidence for treating axial back/lumbar radiculopathy, neuralgia, and complex regional pain syndrome with long-term cost-effectiveness when compared with alternative treatment modalities [7, 8]. It is estimated that over 45,000 SCS procedures have been completed in the USA and over 80,000 worldwidely, with the demand on the uprising.
Several high-quality studies proved that around 50% of the patients could achieve long-term (at least 2 years) 50% pain relief in traditional low-frequency stimulation [9, 10]. Better results were shown in the newly developed stimulation settings such as 10-kHz high-frequency, in which 80% of the patients maintained at least 50% pain relief over 2 years [11]. Continued technological development of SCS (such as electrode specifications, electrode configurations, programming parameters) keeps extending the new indications that used to be refractory to the therapies into the competence circle of physicians [3, 4, 12]. While SCS continues to benefit an increasing number of pain patients in developed areas of the world, there is a small group of chronic pain sufferers that could access to the therapy in China.
There are only over 200 cases (excluding temporary lead implantation) being performed in China. Factors contributing to this situation include poor affordability of patients, ignorance of patients, and a tremendous knowledge gap with only few doctors in the core cities being able to implement this procedure. Thus, RCT evidence is needed to confirm the efficacy and safety of SCS in Chinese chronic intractable pain patients.
Method/design
Type of trial
The CITRIP study is a standard double-blind, placebo-controlled, enriched-enrollment, randomized-withdrawal (EERW) study to evaluate the clinical effectiveness and safety of SCS using G122R implanted pulse generator (IPG; PINS, Inc., Beijing, BJ, China) on chronic intractable trunk or limb pain patients. The EERW design was proposed for the analgesic drugs in chronic noncancer pain [13] and as a well-accepted design for the evaluation of chronic pain (systematic review [14]; several studies using EERW [15,16,17,18]). The main difference between EERW and classic RCT is to shift the point of randomization. The enrichment enrollment of the design shifts our focus to the individual “true” responders (defined as both achieving at least 50% pain intensity reduction and tolerating the treatment for 12 weeks) [14], which is important in the clinical practice.
According to the trial flow chart (Fig. 1), the participants with chronic pain will be screened strictly based on the inclusion and exclusion criteria. Participants will receive the temporary spinal cord stimulation, and the eligible subjects could undertake the implantation of IPG. After a 3-month visit (V6), participants will be randomized (1:1) to one of the two groups: the experimental group (EG, stimulation continued in the withdrawal period) or the control group (CG, stimulation ceased in the withdrawal period). The clinical measurements of the two groups are shown based on Fig. 2. The participants in EG will continue to receive SCS therapy for 7 days, while the participants in CG will temporarily cease the SCS therapy during 1-week withdrawal period, or turn on the device in advance if the VAS score increases to the pre-operative baseline. All outcomes will be calculated by blinded statisticians appointed by the clinical research institute of Peking University following the end of the trial.

Flow chart of the CITRIP study

Standard Protocol Items (SPIRIT) for the CITRIP study
The CITRIP study protocol was written in accordance with the Standard Protocol Items: Recommendations for Interventional Trials Statement (SPIRIT). A SPIRIT checklist is included in Additional file 1. The trial will be carried out according to the principles of the Declaration of Helsinki (Edinburgh version, 2000).
Study setting
The trial will be conducted at approximately 10 investigative centers domestically. Additional centers may be added during the course of the study. Every center should complete at least 4 cases of IPG implantation. No single center will exceed 30% of all enrolled patients. The protocol has been approved by the Clinical Trial Ethics Committee of China-Japan Friendship Hospital (protocol number 2019-135-Q17-2; date January 23, 2019) and registered on ClinicalTrials.gov protocol system (Clinical Trials Identifier: NCT03858790).
Selection of patients
The study population comprises of patients suffering from chronic intractable trunk or limb pain. A patient must meet all of the inclusion criteria and none of the exclusion criteria to be eligible for the study. China has a large population of chronic intractable trunk or limb pain patients. The enrollment is predicted to come to a halt before December 2019.
Inclusion criteria
Inclusion criteria are shown in Table 1.
Exclusion criteria
Exclusion criteria are shown in Table 2.
Interventions
Pain treatment will be evaluated. To confirm the eligibility of the participants, the investigator should review the participant’s history of therapies for pain. Intractable pain means pain that is resistant to pharmacological agents or interventional therapies (for example, nerve block injections, radiofrequency, acupuncture, functional restoration, physical therapy, and psychological interventions, such as cognitive behavioral therapy) or intrathecal drug delivery or surgery.
Participants will undergo an SCS trial period (screening test, 4 days minimum, 15 days maximum). This screening test will be conducted with one or two percutaneous lead(s) or paddle leads. If successful (VAS score improves over 50% compared with baseline), an implanted pulse generator will be implanted. Participants who cannot accept the paresthesia or refuse to proceed the IPG implantation should also be considered as failing the screening test. After implantation, devices will be programmed to the optimal programming parameters and can be adjusted with the patient programmer, within the settings programmed in the clinic. Subjects will be provided with a patient programmer manual and will be instructed on the proper use and handling of the patient programmer.
Implant procedure
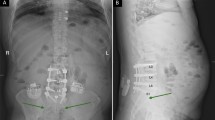
Consistent with the clinical practice, in the trial period, the electrode implantation was undertaken in the awake surgery fashion. The subject was brought to a conscious sedated state while maintaining local anesthesia. An external pulse generator (T802, PINS, Inc., Beijing, BJ, China) was utilized to stimulate the spinal cord using the percutaneous or paddle lead with the guidance of fluoroscopy. The subject should provide verbal feedback regarding paresthesia coverage of painful regions. Lead was always repositioned to achieve better paresthesia overlap of painful areas. Once 50% reduction of pain was attained during the trial period, an IPG implantation surgery was subsequently performed. The leads, anchored to the supraspinous ligaments, were tunneled to the pocket site and connected to the IPG. Intraoperative impedance testing ensured electrical integrity. A subcutaneous pocket was created in the buttock, chest, or abdomen according to the physician and subject’s preference.
Lead programming
After lead implantation, investigators will review the contact combination in a consecutive manner. The paresthesia coverage and related lead configuration, amplitude, pulse width, and frequency will be recorded. The optimal contact combination (usually the best coverage with the lowest output) will be used. After IPG implantation, investigators are recommended to program 4 sets of lead configurations (2 paresthesia sets and 2 paresthesia-free sets) for the participants according to the programming record in the screening phase. The programming will be completed before discharging the hospital. Participants are allowed to change among the predetermined configurations by themselves (switch the presetting configurations or increase/decrease the amplitude within a presetting limit). The following must be underlined during the programming:
-
1.
Programming sessions should not exceed 1 h in case the fatigue leads to the cooperation problem or subjective bias.
-
2.
Paresthesia configuration is firstly recommended for the investigators; paresthesia-free configuration usually requires more frequent recharges.
Remote programming
In China, there are only 200–300 cases of SCS being implanted every year. Only a few doctors in central cities of different areas are trained to implement this surgical technique. China is a large country with 1.3 billion people, most of whom lived in the underdeveloped areas. Based on the remote programming system developed for deep brain stimulation [19, 20], the remote programming function has been well equipped for the SCS system and will be used in this trial to facilitate the post-operative management of participants.
For a short introduction, the remote system could enable the doctors to implement the programming of the SCS system at any time in patients with network coverage. IPG will connect with the patient’s cell phone by the Bluetooth at the same time doctor’s computer connecting with the patient’s cell phone. Real-time video interaction will give doctors sufficient information to adjust the configuration and parameters of the SCS.
In this trial, participants could have access to the doctors via this remote programming system at home or at a local clinic. It provides participants convenience with less expenditure. As for the use of the remote system, several issues should be stressed:
-
1.
All participants and at least one caregiver are ensured to be familiar with the remote programming and be able to cooperate with investigators to complete the whole procedure before leaving the hospital.
-
2.
Participants could apply for the remote programming within V4-V7. Investigators will adjust the configuration and parameters of the SCS based on the initial programming records.
-
3.
If the therapeutic effect is not satisfactory as in the hospital, an on-site programming is recommended.
Randomization and withdrawal
Randomization allocation will be concealed from the investigators and participants and implemented with a central automatic web-based data management system of the third party (CIMS, Ltd., Chengdu, China). Permuted blocks and stratification will be used to generate the randomization assignments, in order to keep the balance of patients in each center. The stratification is based on the medical center to which the participant belongs.
After a 3-month visit, subjects will run into the 7-day withdrawal phase. They will be randomly assigned to the experimental group (EG, turning on the SCS) or control group (CG, turning off the SCS) in a 1:1 ratio, according to the “random number table” generated by the CIMS clinical trial central randomization system of the third party. For the nature of spinal cord stimulation, the trial could not be strictly double-blinded (SCS therapy induces paresthesia). In order to carry out the trial under unbiased conditions with investigators and participants as much as possible, the participant will not be told to which group he/she is assigned. Also, each center designates a specific investigator to access to the randomization system with his own password and account. When a certain subject needs to run into the randomization phase, this investigator enters the subject’s trial number and gets the unique random number of this subject from the randomization center, which assigns the subject to EG or CG. This designated investigator who could access to the randomization system is the only unblinded investigator during the blind phase. He/she is responsible for the turning on/off of the device, modulation of the parameters, and checking whether the VAS score returns to the baseline. Other investigators, particularly the evaluators, will be blind to the treatment conditions until the end of the open-label period. The blinded investigators will be trained to obey the experimental rules and not enquire into the randomized results.
Participants randomized to the EG continued the SCS stimulation-on, while participants in the CG will turn off the SCS during the 7-day withdrawal period. CG participants will turn on the SCS if the VAS score returns to the pre-operative baseline and lasting for 2 days. Otherwise, turning on the SCS is on the 8th day post-randomization.
Primary outcome
The primary outcome will be the difference of maximal visual analog scale (VAS) score between EG and CG in the withdrawal period compared with the baseline in the withdrawal period. The VAS is a subjective evaluation scale for pain rating. Participants will record their trunk and limb pain using a (paper) pain diary every day for a 7-day withdrawal period. For the withdrawal baseline and other follow-up visits, participants will record a 4-day period of VAS score. The proportion of subjects in each group with 50% reduction in average VAS score at the trial period (V2) compared with pre-operative baseline will undertake the IPG implantation and will be calculated.
The primary outcome calculation formula is:
where VASEGbase is the baseline 4-day average VAS score of EG before the withdrawal period, VASEGwd is the maximal VAS score of EG in the withdrawal period, VASCGbase is the baseline 4-day average VAS score of CG before the withdrawal period, and VASCGwd is the maximal VAS score of CG in the withdrawal period.
Secondary outcomes and safety outcomes
The secondary outcomes include VAS score change at 1-, 3-, and 6-month follow-ups; responder rate (VAS score improving by 50%); achievement rate of a desirable pain state (no worse than mild pain, VAS scores ≤ 4) [21]; awake times during sleep; Beck Depression Inventory for depression evaluation [22]; short-form 36 for quality of life evaluation [23]; drug usage; and satisfaction rating of the device (a modified Patients’ Global Impression of Change scale [24], which consists of PGIC items and ratings for the satisfaction for the device).
The occurrence rate of AE/SAE during the different periods of the trial will be used to evaluate the safety of SCS. The investigators are responsible for the management of participants’ AEs and SAEs. The clinician will turn off the SCS therapy and evaluate the vital signs of the participant if the SAE occurs, then the related therapy would be given for the participant. The investigators should determine whether the IPG and/or electrode should be removed or replaced according to the event. All the above events must be reported to the trial manager within 24 h.
Sample size and power calculations
The primary comparison is the difference between EG and CG in terms of VAS score in the withdrawal period. According to the results of the study [25], the clinically important outcome of VAS score is 20 mm at most, which does not vary with gender, age, cause, or severity of pain [26, 27]. A sample size of 48 (24 per arm) achieves 80% power to detect the significant difference using a two-sided, two-sample t test at a significance level (alpha) of 0.025. We assumed the true difference between the means to be 0 and a standard deviation of the outcome of 2.6. Thus, the sample size calculation formula was:
where |D| is the expected mean difference of the two groups, ∆ is the superiority threshold value (0, here), Z is the quantile of the standard normal distribution, α is the first class error level for the statistical test (0.025, here for unilateral test; 0.05 for bilateral test if need), and β is the second class error level for the statistical test (0.2, here). The calculated sample size per group through this calculation is 21. Considering that the maximum possible loss rate is 10%, the total sample size is 24 pairs (N = 48) [24].
These 48 cases will use the G122R rechargeable IPG combined with percutaneous leads (PINS, Inc., Beijing, BJ, China). For the non-rechargeable model G122 and paddle lead, other 6 participants will be recruited for each condition (3 cases for each). No statistical requirements are needed for these 6 cases. In summary, the final sample size is 54 (24 pairs plus extra 6).
To implement within-group comparisons, data will be analyzed with t tests and χ2 tests for continuous variables and categorical variables, respectively. The Kolmogorov-Smirnov test or parametric tests will be performed with the data of normal distributions. Rank-sum test will be used for the non-parametric data. The factors that affect curative effects will be analyzed with a multinomial logistic regression method. A p value < 0.05 will be considered as statistically significant. A validated statistical software package will be used for the analyses of the study results (for example, SAS).
Quality control and trial monitoring
Clinical documents have been formulated before the implementation of the trial with the discussion of the clinical experts from different medical centers, including standard operating procedure (SOP), investigator’s brochure, and detailed research plan. All staff participating in this program have received the meticulously organized training including different parts of patient enrollment, surgery technique, programming demonstration, database demonstration, and CRF completion. All the investigators from every center should complete the consistency training held by China-Japan Friendship Hospital.
As for the assessment, because of the nature of spinal cord stimulation, the trial could not be strictly blinded (SCS therapy induces paresthesia). In order to minimize the bias, participants will fill the pain diary without study staff consultation or visibility. Also, the resulted VAS score is an average score of the consecutive 4-day ratings (except the VAS score in the withdrawal period, which is the maximal score).
To quantify performance bias, the drug treatment and addition/removal of SCS of all the participants are followed during the whole study. The drug usage from the baseline to the end of the study will be compared in order to interpret the between-group outcome differences.
Data is firstly monitored by the investigator himself/herself to ensure the authenticity and completeness. Then the certificated clinical research associate (CRA) will monitor, audit, and ensure the trial is conducted and data are generated, documented (recorded), and transported in compliance with the protocol, GCP, and regulatory requirements. CRA will do the monitoring and quality control of the study on behalf of the sponsors (PINS Medical, Ltd., Beijing, China). Monthly checks will be implemented on the CRF of each participant, data accuracy, protocol compliance, and violation issues. The quality control facility of each center will ensure the study’s quality with the processes of the program usually for 3 times at the first enrollment, half the enrollment, and end of the enrollment.
An independent clinical committee consisting of a minimum of three independent clinicians will review all the adverse events and discuss and determine any relationship to the SCS therapy.
The final report will follow the Consolidated Standards of Reporting Trials (CONSORT) extension guidelines for non-pharmaceutical interventions.
Discussion
The past five decades have witnessed the development of SCS from a conception to a well-established therapy, named as “the last resort therapy” for chronic intractable pain. Numerous studies have demonstrated that SCS is clinically effective for the chronic intractable pain induced by failed back surgery syndrome, complex regional pain syndrome, periphery ischemic pain, refractory angina pectoris, and many other diseases [3,4,5,6, 13]. Here, we propose this first SCS trial in China using a randomized withdrawal design demonstrating that the SCS is well tolerated and effective for the management of chronic intractable pain. The outcome measures selected for this trial are based on the previous RCTs of SCS [10] and IMMPACT recommendations [28, 29].
Double-blind, placebo-controlled, enriched-enrollment, randomized-withdrawal (EERW) design usually has the screening phase, titration phase, and maintenance of effective dose and randomization withdrawal phase [14]. Just like other pain drugs focused on the central or peripheral mechanism of pain, SCS is regarded as the “electroceuticals” to reduce pain with several mechanisms [14, 30, 31]. The programmable and reversible characteristics make it perfectly fit into the EERW study design. In clinical practice, pain doctors usually perform a screening procedure for the patients to test whether the IPG should be implanted (“natural” screening phase, in which participants who get 50% or more reduction of the pain are candidates for the IPG implantation) [32]. Patients will receive a thorough programming to achieve the best pain relieve (titration phase). In order to get the “true responders,” we set the maintenance phase as 3 months to stabilize the therapeutic effect of SCS before randomization. Three months after implantation is usually enough for the parameter settlement and a stable therapeutic effect (maintenance of effective dose). Since almost all the intractable chronic pain patients’ symptoms will return within 24 h after turning off the SCS [33], we choose to have the 7 days of withdrawal period to avoid a carry-over effect in this study. Besides, the randomized withdrawal period may also have contributed to the study sensitivity, as it allowed patients who achieved good pain control to experience a loss of pain control. Sensitivity and awareness of loss of pain control could be a more profound experience than the gain in pain control that occurs with conventional studies. Thus, with the idea of electroceutical and design of EERW, this trial could provide more insights relevant to clinical practice.
In 2018, there have been less than 200 SCS cases (IPG implantation) completed in China. There is a big knowledge gap for the doctors and patients. To implement the SCS trial, a standard training program will be proceeded with the help of China-Japan Friendship Hospital, whose PI is well trained in USA and also a pioneer in this field in China. The sponsor has to make sure that all the investigators are capable of carrying out the trial protocol, especially performing the surgery and post-operative programming.
Besides lack of the expertise, China is a big country with the area of 9,600,000 km2 and people of 1.39 billion. The Gross Domestic Product (GDP) per capita in 2018 is $9376.97, one sixth of US’s. For this development level, post-operative programming is still a big burden for the neuromodulation patients. As for the neuromodulation therapies, surgery is only a beginning, with post-operative management being crucial. In particular, the uneven distribution of medical resources makes it even harder for the patients in the remote areas of China. To solve the challenges above, telemedicine could be used to overcome these healthcare barriers including travel distance, shortage in expertise distribution, growing disability or immobility, and lack of accessibility [34, 35].
Based on the remote programming mode we developed for our deep brain stimulation system [19, 20], SCS has been equipped with the remote programming mode. With the well-developed 4G network infrastructure in China, participants could connect directly to the expertise to receive the pain evaluation and programming. Chronic pain is well suited to telemedicine because it could be visually and vocally assessed. Furthermore, to stabilize the therapeutic effect of SCS, telemedicine is associated with higher patient satisfaction [36] and quality indicators [37].
It is important to note that this study has several limitations. Firstly, all the outcome measurements are patient-reported outcomes from a pain diary. The study could be biased with the placebo effect. Another potential limitation of this trial is the follow-up duration of 6 months. We noted that a longer follow-up for assessing chronic SCS therapy over 1 year remains valuable and helpful to reduce the possible placebo effect.
Trial status
Enrollment of participants into this study started in February 2019 and continues. Target enrollment for this study is 54 participants.
Protocol version
Version 1.2 (November 29, 2018)
Availability of data and materials
Data from this study are unavailable before the end of this trial. Data collected during the course of the research will be kept strictly confidential and only accessed by members of the trial team. All participants will be allocated an identification number, and participant’s details will be stored on a secure database of a third party (CIMS, Ltd., Chengdu, China). The datasets analyzed during the current study are available from the sponsor on reasonable request.
Abbreviations
- SCS:
-
Spinal cord stimulation
- IPG:
-
Implantable pulse generator
- CRF:
-
Case report form
- EG:
-
Experimental group
- CG:
-
Control group
- AE:
-
Adverse event
- SAE:
-
Serious adverse event
- SOP:
-
Standard operating procedure
- CRA:
-
Clinical research associate
References
Gaskin DJ, Richard P. The economic costs of pain in the United States. J Pain. 2012;13:715–24.
Vannemreddy P, Slavin KV. Spinal cord stimulation: current applications for treatment of chronic pain. Anesth Essays Res. 2011;5:20–7.
Chakravarthy K, Richter H, Christo PJ, Williams K, Guan Y. Spinal cord stimulation for treating chronic pain: reviewing preclinical and clinical data on paresthesia-free high-frequency therapy. Neuromodulation. 2018a;21:10–8.
Chakravarthy K, Kent AR, Raza A, Xing F, Kinfe TM. Burst spinal cord stimulation: review of preclinical studies and comments on clinical outcomes. Neuromodulation. 2018b;21:431–9.
Taylor RS. Spinal cord stimulation in complex regional pain syndrome and refractory neuropathic back and leg pain/failed back surgery syndrome: results of a systematic review and meta-analysis. J Pain Symptom Manag. 2006;31:S13–9.
Ubbink DT, Vermeulen H. Spinal cord stimulation for critical leg ischemia: a review of effectiveness and optimal patient selection. J Pain Symptom Manag. 2006;31:S30–5.
Rock AK, Truong H, Park YL, Pilitsis JG. Spinal cord stimulation. Neurosurg Clin N Am. 2019;30:169–94.
Deer TR, Grider JS, Lamer TJ, Pope JE, Falowski S, Hunter CW, Provenzano DA, Slavin KV, Russo M, Carayannopoulos A, et al. A systematic literature review of spine neurostimulation therapies for the treatment of pain. Pain Med. 2020;21(7):1421–32.
North RB, Kidd DH, Farrokhi F, Piantadosi SA. Spinal cord stimulation versus repeated lumbosacral spine surgery for chronic pain: a randomized, controlled trial. Neurosurgery. 2005;56:98–107.
Kumar K, Taylor RS, Jacques L, Eldabe S, Meglio M, Molet J, Thomson S, O'Callaghan J, Eisenberg E, Milbouw G, et al. The effects of spinal cord stimulation in neuropathic pain are sustained: a 24-month follow-up of the prospective randomized controlled multicenter trial of the effectiveness of spinal cord stimulation. Neurosurgery. 2008;63:762–70.
Kapural L, Yu C, Doust MW, Gliner BE, Vallejo R, Sitzman BT, Amirdelfan K, Morgan DM, Yearwood TL, Bundschu R, et al. Comparison of 10-kHz high-frequency and traditional low-frequency spinal cord stimulation for the treatment of chronic back and leg pain: 24-month results from a multicenter, randomized, controlled pivotal trial. Neurosurgery. 2016;79:667–77.
Kapural L, Yu C, Doust MW, Gliner BE, Vallejo R, Sitzman BT, Amirdelfan K, Morgan DM, Brown LL, Yearwood TL, et al. Novel 10-kHz high-frequency therapy (HF10 therapy) is superior to traditional low-frequency spinal cord stimulation for the treatment of chronic back and leg pain: the SENZA-RCT randomized controlled trial. Anesthesiology. 2015;123:851–60.
Vinik AI, Shapiro DY, Rauschkolb C, Lange B, Karcher K, Pennett D, Etropolski MS. A randomized withdrawal, placebo-controlled study evaluating the efficacy and tolerability of tapentadol extended release in patients with chronic painful diabetic peripheral neuropathy. Diabetes Care. 2014;37:2302–9.
Moore RA, Wiffen PJ, Eccleston C, Derry S, Baron R, Bell RF, Furlan AD, Gilron I, Haroutounian S, Katz NP, et al. Systematic review of enriched enrolment, randomised withdrawal trial designs in chronic pain: a new framework for design and reporting. Pain. 2015;156:1382–95.
Gimbel J, Spierings EL, Katz N, Xiang Q, Tzanis E, Finn A. Efficacy and tolerability of buccal buprenorphine in opioid-experienced patients with moderate to severe chronic low back pain: results of a phase 3, enriched enrollment, randomized withdrawal study. Pain. 2016;157:2517–26.
Hewitt DJ, Ho TW, Galer B, Backonja M, Markovitz P, Gammaitoni A, Michelson D, Bolognese J, Alon A, Rosenberg E, et al. Impact of responder definition on the enriched enrollment randomized withdrawal trial design for establishing proof of concept in neuropathic pain. Pain. 2011;152:514–21.
Zakrzewska JM, Palmer J, Ettlin DA, Obermann M, Giblin GM, Morisset V, Tate S, Gunn K. Novel design for a phase IIa placebo-controlled, double-blind randomized withdrawal study to evaluate the safety and efficacy of CNV1014802 in patients with trigeminal neuralgia. Trials. 2013;14:402.
Zakrzewska JM, Palmer J, Morisset V, Giblin GM, Obermann M, Ettlin DA, Cruccu G, Bendtsen L, Estacion M, Derjean D, et al. Safety and efficacy of a Nav1.7 selective sodium channel blocker in patients with trigeminal neuralgia: a double-blind, placebo-controlled, randomised withdrawal phase 2a trial. Lancet Neurol. 2017;16:291–300.
Chen Y, Hao H, Chen H, Tian Y, Li L. The study on a real-time remote monitoring system for Parkinson’s disease patients with deep brain stimulators. Conf Proc IEEE Eng Med Biol Soc. 2014;2014:1358–61.
Chen Y, Hao H, Chen H, Li L. The study on a telemedicine interaction mode for deep brain stimulation postoperative follow-up. Conf Proc IEEE Eng Med Biol Soc. 2015;2015:186–9.
Busse JW, Bartlett SJ, Dougados M, Johnston BC, Guyatt GH, Kirwan JR, Kwoh K, Maxwell LJ, Moore A, Singh JA, et al. Optimal strategies for reporting pain in clinical trials and systematic reviews: recommendations from an OMERACT 12 workshop. J Rheumatol. 2015;42:1962–70.
Beck AT, Steer RA. Internal consistencies of the original and revised Beck Depression Inventory. J Clin Psychol. 1984;40:1365–7.
Jenkinson C, Coulter A, Wright L. Short form 36 (SF36) health survey questionnaire: normative data for adults of working age. BMJ. 1993;306:1437–40.
Guy W. ECDEU assessment manual for psychopharmacology (DHEW Publication No. ADM 76-338). Washington, DC: US Government Printing Office; 1976.
Ostelo RW, de Vet HC. Clinically important outcomes in low back pain. Best Pract Res Clin Rheumatol. 2005;19:593–607.
Kelly AM. The minimum clinically significant difference in visual analogue scale pain score does not differ with severity of pain. Emerg Med J. 2001;18:205–7.
Kelly AM. Does the clinically significant difference in visual analog scale pain scores vary with gender, age, or cause of pain? Acad Emerg Med. 1998;5:1086–90.
Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar JT, Haythornthwaite JA, Jensen MP, Kerns RD, Ader DN, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. J Pain. 2008;9:105–21.
Dworkin RH, Turk DC, Farrar JT, Haythornthwaite JA, Jensen MP, Katz NP, Kerns RD, Stucki G, Allen RR, Bellamy N, et al. Core outcome measures for chronic pain clinical trials: IMMPACT recommendations. Pain. 2005;113:9–19.
Famm K, Litt B, Tracey KJ, Boyden ES, Slaoui M. Drug discovery: a jump-start for electroceuticals. Nature. 2013;496:159–61.
Reardon S. Electroceuticals spark interest. Nature. 2014;511:18.
Kopec JA, Abrahamowicz M, Esdaile JM. Randomized discontinuation trials: utility and efficiency. J Clin Epidemiol. 1993;46:959–71.
Wolter T, Winkelmuller M. Continuous versus intermittent spinal cord stimulation: an analysis of factors influencing clinical efficacy. Neuromodulation. 2012;15:13–9 discussion 20.
Achey M, Aldred JL, Aljehani N, Bloem BR, Biglan KM, Chan P, Cubo E, Dorsey ER, Goetz CG, Guttman M, et al. The past, present, and future of telemedicine for Parkinson’s disease. Mov Disord. 2014;29:871–83.
Jitkritsadakul O, Rajalingam R, Toenjes C, Munhoz RP, Fasano A. Tele-health for patients with deep brain stimulation: the experience of the Ontario Telemedicine Network. Mov Disord. 2018;33:491–2.
Dorsey ER, Voss TS, Shprecher DR, Deuel LM, Beck CA, Gardiner IF, Coles MA, Burns RS, Marshall FJ, Biglan KM. A U.S. survey of patients with Parkinson’s disease: satisfaction with medical care and support groups. Mov Disord. 2010;25:2128–35.
Cheng EM, Swarztrauber K, Siderowf AD, Eisa MS, Lee M, Vassar S, Jacob E, Vickrey BG. Association of specialist involvement and quality of care for Parkinson’s disease. Mov Disord. 2007;22:515–22.
Acknowledgements
We would like to thank the entire team of researchers for their work, professional skills, enthusiasm, and great efforts. This includes all the nurses and staff at all hospitals. Thanks for Wai Kin Daniel Ko’s contribution to the manuscript.
Funding
Funding for this study includes The National Key Research and Development Program of China (2016YFC0105502), National Natural Science Foundation of China (81527901), and Beijing New-star Plan of Science and Technology Program (Z181100006218129).
Author information
Authors and Affiliations
Contributions
All authors have made substantial contributions to the design and concept of this study. YL and PM drafted the paper under the supervision of BfF, JJW, and LmL. All authors critically revised the manuscript. YLa, PM, WT, DlX, KM, RcL, DF, WrD, SL, ZjF, ZyF, YJ, LW, and YLo were involved in the collection and analyses of the data. JJW and BfF will supervise the therapist involved in the study. All authors read and approved the final manuscript. No professional writers have been involved in this manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The CITRIP study received ethical approval from the Clinical Trial Ethics Committee of China-Japan Friendship Hospital, covering all participating sites. Important changes to the protocol will be submitted to the Ethics Committee for review. If any modifications of the protocol are approved, the sponsor and funder should be notified first then the PI will notify the centers. A copy of the revised protocol will be sent to the PI to add to the Investigator Site File. Informed consent will be obtained from all participants in the trial. On the consent form, participants will be asked if they agree to the use of their data or choose to withdraw from the trial. Participants will also be asked for permission for the research team to share relevant data with people from the Universities taking part in the research or from regulatory authorities.
Consent for publication
Consent forms for the trial include consent for publication of results in peer-reviewed journals. The results will be reported at conferences and in peer-reviewed publications. The first publication will be based on data from the participating centers and will be analyzed with statisticians’ supervision. Any formal presentation or publication of data collected from this study will be considered as a joint publication by the participating investigators and will follow the recommendations of the International Committee of Medical Journal Editors.
Competing interests
Primary investigator’s grants in this trial are supported by the sponsor. No additional reporting is allowed until the final report of the trial is completed, with the exception of consent by the sponsor. Information on the sponsor’s and investigator’s publishing policies will be described in their clinical trial contract.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:.
SPIRIT 2013 Checklist: Recommended items to address in this clinical trial protocol and related documents. It is strongly recommended that this checklist is read in conjunction with the SPIRIT 2013 Explanation & Elaboration for important clarification on the items. Amendments to the protocol should be tracked and dated. The SPIRIT checklist is copyrighted by the SPIRIT Group under the Creative Commons “Attribution-NonCommercialNoDerivs 3.0 Unported” license.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lu, Y., Mao, P., Wang, G. et al. Spinal cord stimulation for chronic intractable trunk or limb pain: study protocol for a Chinese multicenter randomized withdrawal trial (CITRIP study). Trials 21, 834 (2020). https://doi.org/10.1186/s13063-020-04768-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-020-04768-3