
Abstract
Background
Community-acquired pneumonia (CAP) remains a leading cause of death worldwide, and hypoalbuminemia is associated with worse outcomes. However, it remains uncertain whether albumin administration could have any beneficial effects. We aim to assess whether the administration of albumin in hypoalbuminemic patients with CAP increases the proportion of clinically stable patients at day 5 compared with the standard of care alone.
Methods
This is a trial protocol for a superiority, non-blinded, multicenter, randomized, phase 3, interventional controlled clinical trial. The primary endpoint will be the proportion of clinical stable patients at day 5 (intention to treat), defined as those with stable vital signs for at least 24 h. The secondary endpoints will be time to clinical stability, duration of intravenous and total antibiotic treatment, length of hospital stay, intensive care unit admission, duration of mechanical ventilation and vasopressor treatment, adverse events, readmission within 30 days, and all-cause mortality. The trial has been approved by the Spanish Medicines and Healthcare Products Regulatory Agency. The investigators commit to publish the data in peer-reviewed journals within a year of the study completion date. Subjects will be recruited from three Spanish hospitals over a planned enrolment period of 2 years. A follow-up visit will be performed 1 month after discharge. We have estimated the need for a sample size of 360 patients at a two-sided 5% alpha-level with a power of 80% based on intention to treat. Eligible participants must be hospitalized, hypoalbuminemic (≤ 30 g/L), non-immunosuppressed, adults, and diagnosed with CAP. They will be randomly assigned (1:1) to receive standard care plus albumin (20 g in 100 mL) every 12 h for 4 days or standard care alone.
Discussion
If this randomized trial confirms the hypothesis, it should lead to a change in current clinical practice for the management of hypoalbuminemic patients with CAP.
Trial registration
European Clinical Trials Database (EudraCT) 2018-003117-18. Registered on 12 April 2019. ClinicalTrials.gov NCT04071041. Registered on 27 August 2019
Similar content being viewed by others


Background
Community-acquired pneumonia (CAP) is an important public health problem [1]. This is underlined by an annual incidence of approximately 6.49 per 1000 inhabitants and a 10-fold rise in certain subgroups, such as the elderly and patients with chronic obstructive pulmonary disease [2]. Lower respiratory tract infection, and CAP in particular, is a significant cause of morbidity and mortality and remains a leading cause of death in high-income countries [3, 4].
Despite a decrease in short-term mortality among adult patients hospitalized with CAP over recent years, about 10% of these patients continue to die [2, 5, 6], with this figure rising to 28% in patients admitted to intensive care units (ICU), even when antibiotic treatment is adequate [7]. CAP has a significantly higher long-term mortality rate than many other conditions [8], occurring in nearly 1 in 3 adults 1 year after hospitalization [2]. CAP may also trigger acute cardiac events, further increasing morbidity, length of hospitalization, and mortality [9, 10]. Unsurprisingly, therefore, the economic burden of CAP is high, with an estimated yearly cost of 10.1 billion euros in Europe [11] and 13.4 billion dollars in the USA [12]. Inpatient care accounts for most of this cost, with length of stay, which is linked to a longer time to clinical stability, being the most significant factor. Longer stays also increase the risks of complications, such as phlebitis, ulcers, adverse drug reactions, and nosocomial infection [13].
The progress of patients hospitalized with CAP is crucial in the first days of therapy. In fact, once clinical stability is achieved, the course of infection is usually favorable [14]. Several studies have demonstrated that an inadequate inflammatory response is a major cause of early treatment failure and mortality [15,16,17]. Hypoalbuminemia also occurs often and is associated with poor outcomes in acutely ill patients, but it is unclear if this association is causal [18, 19]. A meta-analysis of 90 cohorts and 9 prospective controlled studies indicated that hypoalbuminemia was an independent predictor of poor outcome [18]. Hypoalbuminemia at admission is associated with higher mortality in severe sepsis [19] and with worse outcomes in patients with CAP [20,21,22,23], including a prolonged time to clinical stability and a longer hospitalization stay [22].
In humans, albumin accounts for about 50% of all plasma protein, while its molecular structure and concentration make it responsible for about 80% of the intravascular oncotic pressure [24]. Albumin metabolism in hospitalized patients is complex, and the pathophysiology underlying the relationship between plasmatic concentrations and ill-health is poorly understood. We know that stress and infection are associated with hypoalbuminemia, with an increase in albumin clearance, rather than decreased albumin synthesis, being dominant [25, 26]. In fact, a higher rate of albumin synthesis is typically observed in patients in ICU [27] and with acute inflammatory abdominal disease [28], and in healthy volunteers within 3 h after endotoxin administration [29].
It is now well accepted that, besides a role in maintaining colloidal osmotic pressure, albumin has secondary functions that are critical to normalizing many of the inflammatory pathways involved in sepsis [30]. Albumin transports many compounds and is responsible for most drug-protein binding, including antibiotics. Hypoalbuminemia can therefore lead to significant pharmacokinetic variation, such as an increased volume of distribution and an increased clearance of antibiotics [31]. Albumin also exerts specific antioxidant functions due to its multiple ligand-binding capacities, with two thirds of extracellular albumin existing in its reduced form. The reduced cysteine-34 (Cys34) residue accounts for 80% of plasma thiols, allowing albumin to trap reactive oxygen species, nitric oxygen, other reactive nitrogen species, and prostaglandins [32]. Moreover, albumin can bind several bacterial products [33]. In an endotoxin animal model, human albumin treatment was shown to have a dose-dependent protective effect on endothelial dysfunction through the inhibition of inflammatory and oxidative stress pathways [34]. Commercially available albumins differed in some of their antioxidant properties, primarily due to different levels of free Cys34 [35]. Our knowledge of the immunomodulatory effects of albumin is derived from in vitro and animal studies. Albumin enhances the ability of antigen-presenting cells to trigger T cell activation by increasing major histocompatibility complex II and human leukocyte antigen-DR expressions [36]. In fact, by binding prostaglandin E2, albumin was able to restore immune competence in patients with decompensated end-stage cirrhosis [37]. Two recent clinical trials demonstrated improved survival and a reduction in spontaneous bacterial peritonitis and other bacterial infections in cirrhotic patients with ascites [38, 39]. In endotoxemic rats, albumin resuscitation improved ventricular dysfunction by improving myocardial hypoxia and decreasing hypoxia-inducible factor (HIF)-1α expression [40].
To date, studies assessing albumin administration have focused on heterogeneous populations of septic and critically ill patients. A large randomized clinical trial evaluating the effect of volume replacement with human albumin versus crystalloid in 7000 critically patients failed to show a survival benefit, but decreased mortality was shown in a predefined subanalysis of patients with severe sepsis [41]. Consequently, the randomized controlled ALBIOS trial studied the impact of albumin administration in severe sepsis and septic shock, and although this also showed no mortality benefit, a post hoc subanalysis suggested that there was a lower 90-day mortality in the albumin group with septic shock [42]. Several systematic meta-analyses of albumin therapy in sepsis or septic shock have shown trends toward 90-day reductions in mortality associated with septic shock [43, 44], especially when administered within 6 h [45]. Other studies did not find any benefit [46,47,48]. However, it should be noted that these included different populations, comparators, and durations. This evidence led to the weak recommendation and low quality of evidence for using albumin in addition to crystalloids in the initial resuscitation of sepsis and septic shock in the surviving sepsis guidelines [49]. Although two new trials, ARISS (NCT03869385) and ALBIOSS-BAL (NCT03654001), seek to assess the impact of albumin administration in septic shock, no randomized trial has either been conducted or planned to analyze the effect of albumin treatment in patients with CAP.
We designed the current randomized multicenter trial to assess whether standard care plus albumin therapy will increase the proportion of clinically stable patients at day 5 of admission compared to standard care alone in patients with CAP and hypoalbuminemia.
Design
Study design and setting
This publicly funded academic study will be a non-blinded, multicenter, randomized, parallel, phase 3, interventional controlled clinical trial into the superiority of albumin plus standard therapy over standard therapy alone. The trial is sponsored by Dr. Jordi Carratalà—Hospital de Bellvitge—and began on November 14, 2019, and will run to November 2021. Patients will be recruited in three hospitals in Barcelona, Spain (Bellvitge University Hospital, SCIAS Hospital of Barcelona, and the Hospital Sant-Camil-Consorcio Sanitario Garraf). All items from the WHO Trial Registration Data Set have been registered at the publicly accessible databases ClinicalTrials.gov (NCT04071041) and European Clinical Trials Database (EudraCT 2018-003117-18). Informed consent will be obtained from all patients or their next of kin by the principal and subinvestigators.
Study population
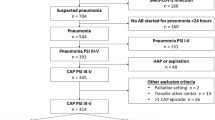
All hospitalized patients with CAP and hypoalbuminemia at presentation will be assessed for eligibility within 24 h of hospital admission.
The inclusion criteria are as follows:
-
Age ≥ 18 years
-
Diagnosis of CAP (Table 1)
-
Serum albumin concentration ≤ 30 g/L at presentation
The exclusion criteria are as follows:
-
Pregnancy or lactation
-
Immunosuppression, such as chemotherapy or radiotherapy (< 90 days), immunosuppressive drugs, corticosteroids within 2 weeks of enrolment (≥ 15 mg/day of prednisone or equivalent), HIV-positive (CD4 count < 200), and solid organ or hematopoietic cell transplant recipients
-
Severe clinical status with expected survival ≤ 24 h
-
Congestive heart failure (New York Heart Association classes 3 or 4)
-
Any contraindication for albumin administration, such as hypersensitivity to albumin
-
Clinical conditions with an indication for albumin (e.g., hepatic cirrhosis with ascites, malabsorption syndrome, or nephrotic syndrome)
-
Absence or impossibility of obtaining informed consent from patient or next of kin
-
Patient already included in another clinical trial for a treatment method
Patient and public involvement
When designing the study, our first priority was patient well-being. However, we have not included either patients or the public in the design, recruitment, or conduct of the study. Participants can request information about the study results through the principal investigator at any time.
Randomization and allocation concealment
The Biostatistics Unit at IDIBELL will generate a centralized pre-specified block randomization list. The program will assign patients on a 1:1 basis to either standard care plus albumin or standard care alone. The block size will be ten, and participants will be stratified by center. The allocation list will be stored at the Biostatistics Unit.
Intervention
Patients in the intervention group will be randomly allocated to receive standard care plus albumin 20% (20 g in 100 mL; Albutein Instituto Grifols, S.A. Can Guasch 2, Parets del Vallès, 08015 Barcelona, Spain) intravenously every 12 h for 4 days or until death, discharge, or clinical stability, whichever occurs first. Patients enrolled in the study will not require extended hospitalization or additional follow-up. Patients will receive empirical antibiotic therapy according to the relevant guidelines, as soon as CAP is confirmed. All microbiological assessments and additional treatments (e.g., oxygen, bronchodilators, corticosteroids, analgesics, vasoactive agents, fluid resuscitation, and mechanical ventilation) will be at the discretion of the treating physicians in both groups. The time of discharge and the duration of antibiotic therapy will not be determined by the study investigators, but by the treating team.
Primary endpoint
The primary endpoint will be the proportion of clinically stable patients at day 5 of hospital admission. Clinical stability will be defined as previously described by Halm et al. as achieving normal oral intake, normal mental status (or usual level of function), and stable vital signs for at least 24 h (Table 2).
Secondary endpoints
The following secondary endpoints will be assessed:
-
Time to clinical stability (days) measured from hospital admission
-
Duration of intravenous and total antibiotic treatment (days)
-
Length of hospital stay (days)
-
ICU admission (time to discharge and durations of treatment with vasopressors and mechanical ventilation)
-
Nosocomial infection during hospitalization
-
Adverse events
-
Hospital readmission within 30 days after discharge
-
All-cause mortality at 5 and 30 days, as well as 30 days of discharge
Stop treatment criteria
Comorbidities of CAP will not be considered sufficient to withdraw a patient from the study. However, the following stop criteria will be applied:
-
Any unexpected serious adverse event between day 0 and day 5
-
Clinical judgment of an attending physician or study investigator
-
Voluntary withdrawal of consent
-
Serious protocol violation
-
Loss to follow-up
-
Pregnancy during the study
-
Congestive heart failure (New York Heart Association classes 3 or 4).
Follow-up and data collection
All included patients will be daily assessed by a member of the investigating team until discharge or death. Consistent with routine clinical practice, a follow-up visit will be arranged for all participating patients 1 month after discharge. For patients who do not attend follow-up or in case of mobility restrictions in response to the COVID-19 pandemic, a structured telephone interview will be done to assess outcomes. A summary of the trial visit schedule and associated assessments is displayed in Fig. 1. Baseline data will include the following: date and time of randomization, demographic data, medical history items, relevant comorbidities, clinical features, causative organisms, antibiotic susceptibilities, antibiotic treatments, other medications, biochemical and microbiological analysis, and variables for the pneumonia severity index (PSI). Albumin concentration will be determined on days 0 and 5. Medication dosages will be recorded, and adverse events will be monitored appropriately throughout the study. In-hospital complications and cardiac events will be assessed. Any additional diagnostic testing in either group will be at the discretion of the treating physician. All data will be recorded on a secure web application used for building and managing online databases (REDCap). The sponsor and investigator will have access to the final trial dataset. Authorized staff may examine the records for quality assurance and audit purposes.

Participant timeline
Statistical analysis plan
Sample size and power calculations
This study is designed to show the superiority of the intervention group (standard care plus albumin) at a two-sided 5% alpha-level with a power of 80% based on intention to treat. According to our own experience, the prevalence of hypoalbuminemia is 37% in patients presenting with CAP, with a predicted mortality rate of 10% [22]. We predicted that 58% of participants would be clinically stable at day 5 in the standard care group, with a difference of 15% between the two treatment arms. Given a possible deviation of 10% from the protocol, we will need a sample of 360 patients (180 per arm) to achieve a statistical power of 80% and a 5% significance level (two-sided). We aim to screen 900 patients.
Type of analysis
We will use IBM SPSS version 21.0 (IBM Corp., Armonk, NY, USA) and R for Windows (R Foundation for Statistical Computing, http://www.r-project.org) for data analysis.
The primary analysis population is the full data set that includes the intention to treat population (all randomized patients). Every effort will be made to minimize the number of losses to follow-up. A secondary analysis will be conducted in a per-protocol population, excluding patients not treated according to the protocol. For sensitivity analysis, the primary analysis will then be repeated in the per-protocol population and by multivariable t test model (adjusted for potentially important confounders, such as age and PSI score).
We also plan to investigate whether the treatment effect varies between different CAP severities, between patients with and without chronic cardiac disease, and between patients with and without bacteremia. We will conduct the pre-specified subgroup analyses by including appropriate interaction terms in the multivariable t test. Complete case analyses will be used for secondary endpoints, for which we will calculate both unadjusted and adjusted estimates of the effect size and corresponding 95% confidence intervals by linear, logistic, or Cox proportional hazards regression.
The trial will be open, but the statistical analyst will be blind to the intervention and control group allocation.
Interim analysis
To ensure sufficient statistical power, the sample size will be recalculated once half of the initial study population (180 patients) has been recruited. The necessity to increase the initial estimated sample size will be considered based on any variabilities encountered.
Monitoring
Data will be monitored by a Data Monitoring Committee from the IDIBELL Clinical Research and Clinical Trials Unit. This committee is co-funded by the European Union and the Instituto de Salud Carlos III, Spanish Ministry of Science, Innovation and Universities, and it is independent of the sponsor.
Adverse event reporting
An independent safety monitoring committee will review safety data. Any adverse event and its possible relationship to the study drug will be noted in patients’ clinical records. All severe adverse events (including death), and any related adverse events, will be recorded on the electronic case report form according to the Common Terminology Criteria for Adverse Events. An adverse event will be considered related to the study medication whenever a temporal relationship suggests a reasonable causal connection and/or the occurrence cannot be better explained by factors like the clinical status or another therapeutic intervention. The investigators will declare any serious adverse events to the sponsor within 24 h, and we will present a yearly security update report to the local regulatory agency consistent with the recommendations of the International Council for Harmonisation (ICH E2F).
Indemnities
This study is classified as a low-intervention trial. According to Spanish law (Real Decreto 1090/2015), all damages incurred should be covered by the civil liability insurance of the participating study centers. An ad hoc insurance for the SCIAS Hospital of Barcelona (a private institution) has been taken out.
Dissemination
The results will be presented at national and international meetings. The investigators commit to publish the data in peer-reviewed journals within a year of the study completion date. The datasets generated during the study will be available from the corresponding author on reasonable request.
Protocol amendments
Relevant protocol modifications will not become effective until approved by the relevant authorities and the Drug Research Ethics Committee. Exceptions are changes to protect patients from imminent harms or exclusively concerning logistic and administrative aspects.
A protocol amendment adding pneumonia caused by SARS-CoV-2 to the exclusion criteria has been submitted and is currently pending decision from the relevant authorities and the Drug Research Ethics Committee.
Discussion
Hypoalbuminemia is associated with worse outcomes in both sepsis and CAP [19,20,21,22,23]. To date, studies evaluating the efficacy of albumin therapy have been performed in heterogeneous populations of critically ill and septic patients. As such, they have rendered conflicting results [41, 42], although some have indicated clear benefits, notably in post hoc subgroup analyses [43]. We hypothesize that albumin administration in hypoalbuminemic patients with CAP could improve both the immune response and degree of organ dysfunction, thereby reducing the length of hospitalization and the time to clinical stability. This open-label randomized trial is designed to test this hypothesis, and it is expected that the results will determine if the addition of albumin therapy to standard care improves clinical outcomes. Even if the results are negative, we can confirm that there is no justification to change current clinical practice. However, if our hypothesis is proven, our study results could lead to a major change in practice.
Trial status
The present manuscript describes the current authorized protocol at submission: version 1.1, 28 March 2019. The first patient was recruited on 14 November 2019, and enrollment is planned to run to November 2021. Recruitment was temporally halted from 14 March 2020 to 7 June 2020 due to the SARS-CoV-2 pandemic.
Availability of data and materials
Not applicable
Abbreviations
- BP:
-
Blood pressure
- CBC:
-
Complete blood count
- CAP:
-
Community-acquired pneumonia
- EudraCT:
-
European Clinical Trial Database
- HIV:
-
Human immunodeficiency virus
- HR:
-
Heart rate
- IDIBELL:
-
Bellvitge Biomedical Research Institute
- ICU:
-
Intensive care unit
- PSI:
-
Pneumonia severity index
- RR:
-
Respiratory rate
- SAE:
-
Serious adverse event
- SO2 :
-
Oxygen saturation
- T°C:
-
Body temperature Celsius
References
Metlay JP, Waterer GW, Long AC, Anzueto A, Brozek J, Crothers K, et al. Diagnosis and treatment of adults with Community-acquired pneumonia. An official clinical practice guideline of the American Thoracic Society and Infectious Diseases Society of America. Am J Respir Crit Care Med. 2019;200:E45–67.
Ramirez JA, Wiemken TL, Peyrani P, Arnold FW, Kelley R, Mattingly WA, et al. Adults hospitalized with pneumonia in the United States: incidence, epidemiology, and mortality. Clin Infect Dis. 2017;65:1806–12.
World Health Organization (WHO): Summary tables of mortality estimates by cause, age and sex, globally and by region, 2000–2016. https://www.who.int/healthinfo/global_burden_disease/estimates/en/. Accessed 28 Oct 2019.
Naghavi M, Abajobir AA, Abbafati C, Abbas KM, Abd-Allah F, Abera SF, et al. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390:1151–10.
Wuerth BA, Bonnewell JP, Wiemken TL, Arnold FW. Trends in pneumonia mortality rates and hospitalizations by organism, United States, 2002–2011. Emerg Infect Dis. 2016;22:1624–7.
Marshall DC, Goodson RJ, Xu Y, Komorowski M, Shalhoub J, Maruthappu M, et al. Trends in mortality from pneumonia in the Europe union: a temporal analysis of the European detailed mortality database between 2001 and 2014. Respir Res. 2018;19:81.
Walden AP, Clarke GM, McKechnie S, Hutton P, Gordon AC, Rello J, et al. Patients with community acquired pneumonia admitted to European intensive care units: an epidemiological survey of the GenOSept cohort. Crit Care. 2014;18:R58.
Eurich DT, Marrie TJ, Minhas-Sandhu JK, Majumdar SR. Ten-year mortality after community-acquired pneumonia. A prospective cohort. Am J Respir Crit Care Med. 2015;192:597–604.
Viasus D, Garcia-Vidal C, Manresa F, Dorca J, Gudiol F, Carratalà J. Risk stratification and prognosis of acute cardiac events in hospitalized adults with community-acquired pneumonia. J Infect. 2013;66:27–33.
Corrales-Medina VF, Musher DM, Wells GA, Chirinos JA, Chen L, Fine MJ. Cardiac complications in patients with community-acquired pneumonia. Circulation. 2012;125:773–81.
Welte T, Torres A, Nathwani D. Clinical and economic burden of community-acquired pneumonia among adults in Europe. Thorax. 2012;67:71–79.
Tong S, Amand C, Kieffer A, Kyaw MH. Trends in healthcare utilization and costs associated with pneumonia in the United States during 2008–2014. BMC Health Serv Res. 2018;18:715.
Hauck K, Zhao X. How dangerous is a day in hospital? A model of adverse events and length of stay for medical inpatients. Med Care. 2011;49:1068–75.
Halm EA, Fine MJ, Marrie TJ, Coley CM, Kapoor WN, Obrosky DS, et al. Time to clinical stability in patients hospitalized with community-acquired pneumonia: implications for practice guidelines. JAMA. 1998;279:1452–7.
Kellum JA, Kong L, Fink MP, Weissfeld LA, Yealy DM, Pinsky MR, et al. Understanding the inflammatory cytokine response in pneumonia and sepsis: results of the Genetic and Inflammatory Markers of Sepsis (GenIMS) Study. Arch Intern Med. 2007;167:1655-63.
Méndez R, Menéndez R, Amara-Elori I, Feced L, Piró A, Ramírez P, et al. Lymphopenic community-acquired pneumonia is associated with a dysregulated immune response and increased severity and mortality. J Infect. 2019;78:423–31.
Siljan WW, Holter JC, Nymo SH, Husebye E, Ueland T, Aukrust P, et al. Cytokine responses, microbial aetiology and short-term outcome in community-acquired pneumonia. Eur J Clin Investig. 2018;48:e12865.
Vincent J-L, Dubois M-J, Navickis RJ, Wilkes MM. Hypoalbuminemia in acute illness: is there a rationale for intervention? A meta-analysis of cohort studies and controlled trials. Ann Surg. 2003;237:319–34.
Yin M, Si L, Qin W, Li C, Zhang J, Yang H, et al. Predictive value of serum albumin level for the prognosis of severe sepsis without exogenous human albumin administration: a prospective cohort study. J Intensive Care Med. 2018;33:687–94.
Miyazaki H, Nagata N, Akagi T, Takeda S, Harada T, Ushijima S, et al. Comprehensive analysis of prognostic factors in hospitalized patients with pneumonia occurring outside hospital: serum albumin is not less important than pneumonia severity assessment scale. J Infect Chemother. 2018;24:602–9.
Hatipoğlu U, Wells BJ, Chagin K, Joshi D, Milinovich A, Rothberg MB. Predicting 30-day all-cause readmission risk for subjects admitted with pneumonia at the point of care. Respir Care. 2018;63:43–49.
Viasus D, Garcia-Vidal C, Simonetti A, Manresa F, Dorca J, Gudiol F, et al. Prognostic value of serum albumin levels in hospitalized adults with community-acquired pneumonia. J Infect. 2013;66:415–23.
Lee JH, Kim J, Kim K, Jo YH, Rhee J, Kim TY, et al. Albumin and C-reactive protein have prognostic significance in patients with community-acquired pneumonia. J Crit Care. 2011;26:287–94.
Fanali G, Di Masi A, Trezza V, Marino M, Fasano M, Ascenzi P. Human serum albumin: from bench to bedside. Mol Aspects Med. 2012;33:209–90.
Levitt DG, Levitt MD. Human serum albumin homeostasis: a new look at the roles of synthesis, catabolism, renal and gastrointestinal excretion, and the clinical value of serum albumin measurements. Int J Gen Med. 2016;9:229–55.
Ballmer PE, Ochsenbein AF, Schütz-Hofmann S. Transcapillary escape rate of albumin positively correlates with plasma albumin concentration in acute but not in chronic inflammatory disease. Metabolism. 1994;43:697–705.
Barle H, Gamrin L, Essén P, McNurlan MA, Garlick PJ, Wernerman J. Growth hormone does not affect albumin synthesis in the critically ill. Intensive Care Med. 2001;27:836–43.
Komáromi A, Estenberg U, Hammarqvist F, Rooyackers O, Wernerman J, Norberg Å. Simultaneous assessment of the synthesis rate and transcapillary escape rate of albumin in inflammation and surgery. Crit Care. 2016;20:370.
Barle H, Januszkiewicz A, Hållström L, Essén P, McNurlan MA, Garlick PJ, et al. Albumin synthesis in humans increases immediately following the administration of endotoxin. Clin Sci (Lond). 2002;103:525–31.
Ferrer R, Mateu X, Maseda E, Yébenes JC, Aldecoa C, De Haro C, et al. Non-oncotic properties of albumin. A multidisciplinary vision about the implications for critically ill patients. Expert Rev Clin Pharmacol. 2018;11:125–37.
Ulldemolins M, Roberts JA, Rello J, Paterson DL, Lipman J. The effects of hypoalbuminaemia on optimizing antibacterial dosing in critically ill patients. Clin Pharmacokinet. 2011;50:99–110.
Taverna M, Marie AL, Mira JP, Guidet B. Specific antioxidant properties of human serum albumin. Ann Intensive Care. 2013;3:4.
Jürgens G, Müller M, Garidel P, Koch MHJ, Nakakubo H, Blume A, et al. Investigation into the interaction of recombinant human serum albumin with Re-lipopolysaccharide and lipid A. J Endotoxin Res. 2002;8:115–26.
Kremer H, Baron-Menguy C, Tesse A, Gallois Y, Mercat A, Henrion D, et al. Human serum albumin improves endothelial dysfunction and survival during experimental endotoxemia: concentration-dependent properties. Crit Care Med. 2011;39:1414–22.
Plantier J-L, Duretz V, Devos V, Urbain R, Jorieux S. Comparison of antioxidant properties of different therapeutic albumin preparations. Biologicals. 2016;44:226–33.
Aubin É, Roberge C, Lemieux R, Bazin R. Immunomodulatory effects of therapeutic preparations of human albumin. Vox Sang. 2011;101:131–7.
O’Brien AJ, Fullerton JN, Massey KA, Auld G, Sewell G, James S, et al. Immunosuppression in acutely decompensated cirrhosis is mediated by prostaglandin E2. Nat Med. 2014;20:518–23.
Caraceni P, Riggio O, Angeli P, Alessandria C, Neri S, Foschi FG, et al. Long-term albumin administration in decompensated cirrhosis (ANSWER): an open-label randomised trial. Lancet. 2018;391:2417–29.
Di Pascoli M, Fasolato S, Piano S, Bolognesi M, Angeli P. Long-term administration of human albumin improves survival in patients with cirrhosis and refractory ascites. Liver Int. 2019;39:98–105.
Tokunaga C, Bateman RM, Boyd J, Wang Y, Russell JA, Walley KR. Albumin resuscitation improves ventricular contractility and myocardial tissue oxygenation in rat endotoxemia. Crit Care Med. 2007;35:1341–7.
Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R, et al. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med. 2004;350:2247–56.
Caironi P, Tognoni G, Masson S, Fumagalli R, Pesenti A, Romero M, et al. Albumin replacement in patients with severe sepsis or septic shock. N Engl J Med. 2014;370:1412–21.
Xu JY, Chen QH, Xie JF, Pan C, Liu SQ, Huang LW, et al. Comparison of the effects of albumin and crystalloid on mortality in adult patients with severe sepsis and septic shock: a meta-analysis of randomized clinical trials. Crit Care. 2014;18:702.
Patel A, Laffan MA, Waheed U, Brett SJ. Randomised trials of human albumin for adults with sepsis: systematic review and meta-analysis with trial sequential analysis of all-cause mortality. BMJ. 2014;349:g4561.
Jiang L, Jiang S, Zhang M, Zheng Z, Ma Y. Albumin versus other fluids for fluid resuscitation in patients with sepsis: a meta-analysis. PLoS One. 2014;9:e114666.
Rochwerg B, Alhazzani W, Gibson A, Ribic CM, Sindi A, Heels-Ansdell D, et al. Fluid type and the use of renal replacement therapy in sepsis: a systematic review and network meta-analysis. Intensive Care Med. 2015;41:1561–71.
Delaney AP, Dan A, McCaffrey J, Finfer S. The role of albumin as a resuscitation fluid for patients with sepsis: a systematic review and meta-analysis. Crit Care Med. 2011;39:386–91.
Uhlig C, Silva PL, Deckert S, Schmitt J, de Abreu MG. Albumin versus crystalloid solutions in patients with the acute respiratory distress syndrome: a systematic review and meta-analysis. Crit Care. 2014;18:R10.
Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving Sepsis Campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. 2017;43:304–77.
Acknowledgements
This study is supported by research grants from the Instituto de Salud Carlos III, Spanish Ministry of Science, Innovation and Universities, through the 2018 call of pre-doctoral contract for training in health research (PFIS contract FI18/00183), with additional help from the European Social Fund (programming period 2014-2020) “The European Social Funds invests in your future,” and grant FIS PI17/01332, co-financed by the European Development Regional Fund “A Way to Achieve Europe.” The project is also supported by Plan Nacional de I+D+i 2013-2016 and Instituto de Salud Carlos III, Subdirección General de Redes y Centros de Investigación Cooperativa, Ministerio de Economía, Industria y Competitividad, Spanish Network for Research in Infectious Diseases (REIPI RD16/0016/0005) - co-financed by the European Development Regional Fund “A Way to Achieve Europe”, Operative program Intelligent Growth 2014-2020. We thank CERCA Programme/Generalitat de Catalunya for institutional support.
Funding
This research project is funded through a public grant obtained from the Instituto de Salud Carlos III, Spanish Ministry of Science, Innovation and Universities (PI17/01332). The funders had no role in the design of this study and will have no input in its conduct; in the acquisition, analysis, or interpretation of data; or in the development and publishing of the manuscript.
Author information
Authors and Affiliations
Contributions
JC, CG, AR AFS, and SV contributed to the concept and design of the trial. The inclusion, data collection, and interpretation will be performed by JC, CG, AR, GA, AFS, GV, YM, LO, MC, and YR. CT developed the statistical analysis plan and calculated the sample size. AR, GA, JC, CG, CT, and JN participated in the writing and submission for publication. No professional writers have been involved. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval was obtained by the Clinical Research Ethics Committee of Bellvitge (reference number HUB-INF-ALBUCAP-402) and the Spanish Medicines and Medical Devices Agency (AEMPS) and is valid for all participating centers (all within in the same autonomous community). Informed consent will be obtained from all patients or their next of kin by the principal investigators. The study will be carried out in accordance with current Spanish (Real Decreto 1090/2015) and European legislation (Regulation 536/2014) and will follow the principles of the Helsinki Declaration (ISO 14155:2011).
Consent for publication
Not applicable
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rombauts, A., Abelenda-Alonso, G., Simonetti, A.F. et al. Effect of albumin administration on outcomes in hypoalbuminemic patients hospitalized with community-acquired pneumonia (ALBUCAP): a prospective, randomized, phase III clinical controlled trial—a trial protocol. Trials 21, 727 (2020). https://doi.org/10.1186/s13063-020-04627-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-020-04627-1