
Abstract
Background
Exotoxins are important virulence factors in Staphylococcus aureus. Clindamycin, a protein synthesis inhibitor antibiotic, is thought to limit exotoxin production and improve outcomes in severe S. aureus infections. However, randomised prospective data to support this are lacking.
Methods
An open-label, multicentre, randomised controlled trial (RCT) will compare outcome differences in severe S. aureus infection between standard treatment (flucloxacillin/cefazolin in methicillin-susceptible S. aureus; and vancomycin/daptomycin in methicillin-resistant S. aureus) and standard treatment plus an additional clindamycin given for 7 days. We will include a minimum of 60 participants (both adult and children) in the pilot study. Participants will be enrolled within 72 h of an index culture. Severe infections will include septic shock, necrotising pneumonia, or multifocal and non-contiguous skin and soft tissue/osteoarticular infections. Individuals who are immunosuppressed, moribund, with current severe diarrhoea or Clostridiodes difficile infection, pregnant, and those with anaphylaxis to β-lactams or lincosamides will be excluded.
The primary outcomes measure is the number of days alive and free (1 or 0) of systemic inflammatory response syndrome (SIRS) within the first 14 days post randomisation. The secondary outcomes measure will include all-cause mortality at 14, 42, and 90 days, time to resolution of SIRS, proportion with microbiological treatment failure, and rate of change of C-reactive protein over time. Impacts of inducible clindamycin resistance, strain types, methicillin susceptibility, and presence of various exotoxins will also be analysed.
Discussion
This study will assess the effect of adjunctive clindamycin on patient-centred outcomes in severe, toxin-mediated S. aureus infections. The pilot study will provide feasibility for a much larger RCT.
Trial registration
Australian New Zealand Clinical Trials Registry, ACTRN12617001416381p. Registered on 6 October 2017.
Similar content being viewed by others


Background
Staphylococcus aureus is a ubiquitous bacterium, colonising at least 50% of the population (20% persistently and 30% intermittently) [1, 2] and causing a wide range of infections. S. aureus bacteraemia (SAB) is associated with a mortality rate of 20–30% in adults [3, 4] and approximately 5% in children [5]. Necrotising pneumonia and severe toxin-mediated infections with S. aureus, however, are associated with a much higher mortality [6,7,8]. This high mortality rate remains despite better understanding of S. aureus disease management in recent decades [8,9,10,11,12,13].
Anti-staphylococcal β-lactams are the drugs of choice for treatment of methicillin-susceptible S. aureus (MSSA) infections, whereas vancomycin (or daptomycin) is the drug of choice for methicillin-resistant S. aureus (MRSA) infections. Successful source control and early effective antibiotics are the crucial factors for treatment success. Use of a single antimicrobial agent is usually recommended, but two- or three-drug combination therapy is advised in selected cases [12, 13], for instance in prosthetic valve infective endocarditis and prosthetic joint infections. The rationale is either for synergy or biofilm penetration. Additionally, several guidelines [12, 14,15,16] recommend adding clindamycin to standard therapy in suspected toxin-mediated S. aureus infections. These recommendations are based on expert opinions with limited clinical evidence available. Clindamycin is a protein synthesis inhibitor (PSI) antibiotic. Although in vitro animal studies and observational human data suggest a possible benefit of adjunctive clindamycin [17], there are no published or registered randomised controlled trials (RCTs) testing this strategy in S. aureus infections.
Staphylococcus aureus toxins
S. aureus virulence is in part due to its exotoxins, surface virulence factors, and enzymes [18]. All S. aureus strains may produce haemolysins, nucleases, proteases, lipases, hyaluronidase, and collagenase. Many strains also harbour genes for toxic shock syndrome toxin-1 (TSST-1), exfoliative toxins, enterotoxins, and leucocidins [19,20,21,22,23]. In an analysis of 429 S. aureus isolates from Germany in 2003 [21] (219 blood isolates and 210 anterior nares isolates, 94% MSSA), 73% of the isolates harboured toxin-related genes. Enterotoxin-G (seg), enterotoxin-I (sei), and toxic shock syndrome toxin (tst) genes were the three most common. In a study by Peacock et al. in the UK in 2002 [23], α-haemolysin was near universal in S. aureus isolates (both carriage and invasive). Other common toxins were β-haemolysin, δ-haemolysin, γ-haemolysin, enterotoxin-G (seg), and enterotoxin-I (sei). Twenty-five per cent of carriage isolates and 30% of bacteraemic isolates had the tst gene detected.
Panton-Valentine leucocidin (PVL) is a S. aureus leukotoxin. Its prevalence varies by population demographics, strain type, and source of infection [24, 25]. PVL-expressing strains have been associated with skin and soft tissue infections (SSTI), which often require surgical intervention [26]. Multi-locus sequence typing (MLST) ST93 S. aureus is a virulent strain, and is common in Australia. It carries lukSF-PV and hyper-expresses α-toxin [27]. This is true for ST93 MRSA, but MSSA of the same sequence type often co-exists [28].
S. aureus virulence is due to multiple determinants, and hence targeting a single factor is unlikely to prove successful in improving disease outcomes [23, 29, 30]. Clindamycin, through its role as a PSI, is expected to limit expression of multiple exotoxins and hence may improve clinical outcomes, but randomised prospective data to support this are lacking.
Antibiotics which act as protein synthesis inhibitors
Certain antibiotics exert their anti-bacterial activity by selectively blocking prokaryotic ribosomal protein synthesis [31, 32]. The antibiotics targeting the 30S ribosomal subunit (aminoglycosides, tetracyclines, and tigecycline) interfere with the principal function of translating mRNA to peptides. The 50S ribosomal subunit acting antibiotics (clindamycin, linezolid, macrolides, streptogramins, chloramphenicol, and fusidic acid) interfere with peptide bond formation [31]. Specifically, MLSB antibiotics (macrolides, lincosamides, streptogramin B) bind to the narrowest portion of the 50S ribosomal subunit tunnel, which has a regulatory function in peptide synthesis [31].
Of the PSI antibiotics, clindamycin has well-recognised anti-toxin activity. It has shown more consistent toxin suppressive activity than other PSI antibiotics [17] and has been recommended in selected toxin-mediated staphylococcal and streptococcal infections. Linezolid has also been reviewed as an anti-toxin agent [33], but its use is limited by cost, potential for haematological toxicity, and preservation for treatment of resistant Gram-positive organisms and mycobacterial infections.
Clindamycin compared to β-lactam antibiotics for anti-toxin therapy
β-lactam antibiotics (penicillins, cephalosporins), the drugs of choice for MSSA infections, are less efficacious in high-inoculum infections. Penicillin has been demonstrated as ineffective against bacteria in a stationary phase of growth (i.e. not actively dividing) [34, 35]. Penicillin treatment (including anti-staphylococcal penicillins) may also stimulate toxin production [36,37,38,39,40]. On the other hand, clindamycin is not affected by the phase of growth or inoculum size [29, 35]. It also represses penicillin-induced exotoxin production [40]. Vancomycin and daptomycin appear neither to induce nor inhibit exotoxin production [39, 41].
Objective
To determine the feasibility of and refine the study design for a future definitive RCT assessing the effect of adjunctive clindamycin on clinical outcomes in children and adults with severe S. aureus infections (both MSSA and MRSA).
Hypothesis
Clindamycin will lead to more rapid resolution of systemic inflammation due to blockade of exotoxin production by S. aureus.
Methods
Study setting
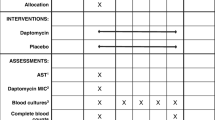
The CASSETTE study is an investigator-initiated, open-label, parallel-group, superiority, multicentre RCT (Fig. 1). This study will be conducted across 12 hospitals in Australia (see Additional file 1: Table S7). Sites are selected based on: prevalence of S. aureus infection (an estimate of at least 10 potentially eligible cases per year for adults and five for children); availability of a committed site principal investigator (PI); and a second person available at each site to assist the PI, either a research nurse, registrar, or physician colleague.

Flowchart overview of the trial design. IV intravenous, MRSA methicillin-resistant Staphylococcus aureus, MSSA methicillin-susceptible Staphylococcus aureus, SIRS systemic inflammatory response syndrome. SPIRIT checklist (2013) provided in Additional file 2
Inclusion criteria
-
1.
Age—both children and adults will be eligible. Infants must have a corrected age of ≥ 28 days.
-
2.
S. aureus (MSSA or MRSA) identified in at least one clinically relevant specimen (including an isolation in a polymicrobial culture when the other isolates are thought to be non-significant).
A “clinically relevant specimen” refers to any specimen where the site PI judges the isolate to be contributing to the patient’s clinical syndrome. This does not have to be a sterile site. These specimens may include blood, fluids from other normally sterile sites (such as joint fluid or cerebrospinal fluid), pus, tissue, or bone obtained surgically. The site PI may additionally make a clinical judgement on whether other specimens from non-sterile sample types yielding S. aureus, such as sputum and superficial swabs, are considered clinically relevant.
-
3.
Ability to be randomised within 72 h of the collection of the index culture.
The index culture is the first clinically relevant specimen collected both after the participant has arrived at a hospital and after the onset of systemic symptoms. If more than one specimen is collected within the same 4-h period, the most clinically significant specimen will count as the index specimen (sterile site isolates are prioritised over others). Randomisation will be performed using a web-based system (REDCap).
Antimicrobial therapy will be clinician determined prior to randomisation. This may include clindamycin and a combination antibiotic that may include coverage for both MSSA and MRSA. The pre-randomisation treatments will be recorded and adjusted for in the analysis.
-
4.
Probability of remaining as an inpatient of the study site hospital for at least 7 days following randomisation (or accessible for follow-up by the site PI, e.g. hospital in the home).
-
5.
Index culture drawn no later than 48 h after hospital admission.
-
6.
Severe disease: evidence of ≥ 2 systemic inflammatory response syndrome (SIRS) criteria within 48 h prior to randomisation and at least one of the following three conditions.
-
6.1.
Septic shock (including Staphylococcal toxic shock syndrome).
-
6.1.
In adults, septic shock is defined as:
-
i.
Mean arterial pressure < 70 mmHg or systolic blood pressure < 90 mmHg despite at least 3 L of fluid administration; or
-
ii.
the need for intravenous vasopressors to maintain organ perfusion (i.e. any receipt of adrenaline, noradrenaline, dopamine, dobutamine, vasopressin, or terlipressin).
In children, septic shock is defined as sepsis with cardiovascular dysfunction. Sepsis is SIRS in a setting of suspected or proven infection. Cardiovascular dysfunction is defined by the International Paediatric Sepsis Consensus Conference [42] as follows—despite administration of isotonic intravenous fluid bolus ≥40 ml/kg in 1 h:
-
i.
decrease in blood pressure (BP) (hypotension) < 5th percentile for age or systolic BP < 2 SD below normal for age (Table 1); or
-
ii.
need for vasoactive drug to maintain BP in normal range (dopamine > 5 μg/kg/min or dobutamine, epinephrine, or norepinephrine at any dose); or
-
iii.
two of the following:
-
a.
unexplained metabolic acidosis: base deficit > 5.0 mEq/L;
-
b.
increased arterial lactate > 2 times upper limit of normal;
-
c.
oliguria: urine output < 0.5 ml/kg/h;
-
d.
prolonged capillary refill: > 5 s; and
-
e.
core to peripheral temperature gap > 3 °C.
-
a.
-
6.2.
Necrotising lung/pleural space infection
Either:
-
i.
an exudative pleural fluid aspirate and has Gram-positive cocci in clusters on Gram stain (in the presence of MSSA or MRSA from another clinically relevant specimen) or has grown S. aureus from the pleural fluid; or
-
ii.
pneumonia which is:
-
a.
multifocal (within the lung); and
-
b.
S. aureus grown from any of sputum, endotracheal aspirate, bronchial lavage, blood, or pleural fluid.
-
a.
The diagnosis of necrotising pneumonia may be aided by computerised tomography (CT) scan, if available.
-
6.3.
Complicated skin/soft tissue/osteoarticular infection which is multifocal and non-contiguous
This includes pyomyositis, septic arthritis, osteomyelitis, skin or soft tissue abscess, or carbuncle.
Non-contiguous multifocal means it is at more than one anatomical site, which is likely to be due to systemic spread rather than direct extension. For example, osteomyelitis of the distal tibia with septic arthritis of the ankle joint does not qualify, as it is contiguous. Lumbar vertebral osteomyelitis with direct extension into the psoas muscle does not qualify. Humeral osteomyelitis with psoas abscess does qualify.
Exclusion criteria
-
1.
Previous severe allergic reaction to both flucloxacillin and cefazolin (for MSSA), or to both vancomycin and daptomycin (for MRSA), or to lincosamides.
-
2.
Previous participation in the trial.
-
3.
Known pregnancy.
-
4.
Currently receiving a lincosamide or other potentially anti-toxin antibiotic which cannot be ceased or substituted (listed in the earlier section “Antibiotics which act as protein synthesis inhibitors”).
-
5.
Participant’s primary clinician unwilling to enrol patient.
-
6.
Moribund (expected to die in next 24 h with or without treatment).
-
7.
Treatment limitations which preclude the use of antibiotics.
-
8.
Significant immunosuppression (prednisolone > 0.5 mg/kg/day for ≥14 days in the last 30 days, other immunosuppressive medication, known HIV with CD4 count < 200, congenital immunodeficiency).
-
9.
Necrotising fasciitis.
-
10.
Clostridiodes difficile-associated diarrhoea or severe diarrhoea (> 6 stools per day or clinician-determined severe diarrhoea in children) from any cause.
Interventions
Control group
The control group will receive “standard therapy”, also called “backbone therapy”. This will consist of IV flucloxacillin 50 mg/kg/dose up to 2 g every 4–6 h given intermittently or via 24-h infusion for MSSA infections. Flucloxacillin can be substituted with IV cefazolin 50 mg/kg/dose up to 2 g every 6–8 h if there is a history of minor allergic reaction to penicillin, defined as rash or unclear allergic reaction, but not anaphylaxis or angioedema, or at the site PI’s discretion. The standard therapy for MRSA infection will consist of IV vancomycin with a loading dose of 25 mg/kg in adults (clinician discretion regarding loading dose in children < 18 years) followed by maintenance dose of 15–20 mg/kg every 12 h (for adults) or 15 mg/kg/dose every 6 h (for children) with subsequent adjustment to maintain trough levels at 15–20 mg/dl, which may require an infusion at clinician discretion or IV daptomycin 6–10 mg/kg/day. Paediatric daptomycin dosing guidance [43]: for 1–6 years old, 12 mg/kg/dose every 24 h; for 7–11 years old, 9 mg/kg/dose every 24 h; and for 12–17 years old, 7 mg/kg/dose every 24 h. The dose of backbone therapy will be adjusted for renal function (Additional file 1: Tables S1–S5).
The duration of standard therapy is clinician determined but should be in line with recommendations in the current version of Therapeutic Guidelines (TG) Antibiotic [14] in adults and with expert consensus in children. The choice between vancomycin and daptomycin, and flucloxacillin and cefazolin is clinician determined, and may be based on local practice or the minimum inhibitory concentration (MIC) of the MRSA isolate against vancomycin.
The usual recommended duration of a standard treatment of S. aureus sepsis in adults is 14–42 days. In children, early switch (after 7–14 days of IV therapy) to oral therapy is practised commonly and is permitted in this trial.
Combination therapy group
All participants assigned to the combination group will receive standard therapy as above. In addition, they will also receive open-label clindamycin (10 mg/kg/dose up to 600 mg QID IV for both adult and children) for 7 days from randomisation (day 1 being the day of randomisation). The intravenous route for clindamycin is recommended but not mandated for the entire duration of therapy (7 days). If a switch to oral therapy is thought to be necessary and appropriate by the site PI, transition to oral clindamycin 450 mg TDS for adults or 10 mg/kg/dose PO TDS (maximum 450 mg) for children is recommended.
Criteria for discontinuing or modifying allocated interventions
-
a.
Adjustment for renal impairment.
Dosage adjustment for the standard therapy medication is as per the recommendations in TG [14] and antimicrobial dosing in renal impairment guidelines of the Children’s Hospital [44] at Westmead, Sydney, Australia (Additional file 1: Tables S1–S5; also includes vancomycin therapeutic drug monitoring). Continuous infusion of the backbone antibiotics (flucloxacillin/cefazolin/vancomycin) during inpatient admission is not routine practice in Australia; however, such use will not constitute a protocol violation. Clindamycin (IV or PO) does not need dose modification in renal impairment.
-
b.
Change of primary standard therapy after randomisation.
The change of primary standard therapy between flucloxacillin and cefazolin (for MSSA) or between vancomycin and daptomycin (for MRSA) is permitted at the discretion of treating clinicians, or if there are issues with supply or stock of these antimicrobials. Any unnecessary changes will be discouraged. Clinicians may change the backbone therapy if the patient develops an adverse drug reaction, such as rash with flucloxacillin or vancomycin, or raised creatinine kinase with daptomycin, or if the vancomycin MIC of the MRSA isolate is found to be ≥ 2 μg/ml, or in cases of persistent bacteraemia whilst receiving vancomycin.
The change within 7 days of randomisation will still be counted as β-lactam therapy and will not violate per-protocol analysis. However, it will affect the subgroup analysis (flucloxacillin vs cefazolin; vancomycin vs daptomycin). Participants who receive the majority of one or the other drugs in the 7 days will be analysed in the respective groups.
-
c.
Clindamycin use after day 7 post randomisation.
The use of clindamycin after the completion of 7 days of randomisation will be discouraged for up to 90 days post randomisation. The treating team will be advised to use an alternative to clindamycin if such treatment is necessary.
Strategies to improve adherence to the protocol, and any procedures for monitoring adherence
-
a.
Training of site investigators.
Each site will have a PI who will be trained in the study protocol, standard operating procedures, and their reporting requirements. The project manager will have regular contact (email or phone) with all enrolling site PIs.
-
b.
Documentation in patient’s medical records.
On day 1 of randomisation, the patient’s enrolment in the CASSETTE trial will be documented in the medical record, and the treating team will be made aware of the recruitment. Study information will be placed in the patient’s folder. The PI or their associates (research nurse or registrar) will check drug charts (paper and/or electronic) daily (except weekends and public holidays) during the period of the intervention. Charts will be reviewed before weekends and public holidays to ensure appropriate doses are prescribed for the period, and to check when clindamycin requires ceasing (i.e. will complete 7 days). Follow-up will be done on the first working day.
-
c.
Monitoring.
As this pilot study is determining feasibility as well as refining assumptions and study design in preparation for a definitive RCT, we intend to utilise off-site risk-based monitoring to be completed by the central coordinating office. The web-based database will have logic checks included in the design to avoid data-entry errors. The central coordinator will also monitor data entry at each site for completeness and data checks will be regularly conducted to ensure protocol compliance.
Relevant concomitant care and interventions that are permitted or prohibited during the trial
Apart from the use of clindamycin, the patient’s management will be at the discretion of the caring team in consultation with the site PI. Procedures undertaken and directly relevant to the study will be recorded (e.g. debridement of infected necrotic tissue, washout of an infected joint, or use of intravenous immunoglobulin). Actions that clearly violate the study protocol, such as early cessation of clindamycin or use of an additional PSI antibiotic, will be discouraged. Site investigators will actively recommend therapeutic drug monitoring or dose adjustment for renal failure per protocol as necessary.
Outcomes
Primary outcome
The primary outcome is the number of days alive and free of SIRS within the first 14 days post randomisation. “Free of SIRS” is defined as meeting < 2 SIRS (i.e. 1 or 0) criteria simultaneously. The day of randomisation is counted as day 1 (not day 0). We used SIRS criteria as they are routinely collected and are a more conservative measure of patient recovery. Also, this was thought to be a single measure of sepsis that could be used in both children and adults.
SIRS criteria in adults [45]
-
1.
Abnormal body temperature (< 36 or 38 °C).
-
2.
Tachypnoea or mechanical ventilation (RR > 20 breaths/min in an adult, age dependent in children).
-
3.
Tachycardia (HR > 90 beats/min in an adult, age dependent in children).
-
4.
Abnormal leucocyte count, > 12 × 109/L or < 4 × 109/L (using last observation carried forward from any FBC).
SIRS criteria in children (Table 1)
SIRS is defined as the presence of at least two of the following four criteria, one of which must be abnormal temperature or leucocyte count. The following are modified from the NSW Clinical Excellence Commission Sepsis Kills guidelines [46] and the International Paediatric Sepsis Consensus Conference [42] held in 2002. Heart rate, respiratory rate, and temperature will be recorded at least daily for the first 7 days, and after this time will be recorded at least daily as long as the patient remains in hospital or hospital in the home (HITH).
-
1.
Abnormal body temperature of < 36 or > 38 °C.
-
2.
Tachycardia or bradycardia (mean heart rate) that is otherwise unexplained and persists over at least 30 min.
-
3.
Tachypnoea (see Table 1) or mechanical ventilation not related to underlying neuromuscular disease or the receipt of general anaesthesia.
-
4.
Abnormal leucocyte count, or > 10% circulating immature neutrophils (“band forms”).
SIRS is considered present if ≥ 2 criteria were met at the same time on any given calendar day. For example: an adult had a heart rate of 95 beats/min at 9 a.m. but no other criteria at this time. She then had a heart rate < 90 beats/min for the rest of the day. At 3 p.m. she had a respiratory rate of 22 breaths/min for 1 h which also then normalised. Although she had two SIRS criteria on the same calendar day, they were not simultaneous and hence she was deemed to be free of SIRS on that day.
If the patient has been discharged from both hospital and HITH before day 14, then SIRS will be assumed to have resolved at the time of discharge if it had not already done so.
White blood counts will be measured on days 1, 3, 5, 7, and 14. There is a ± 1-day window if the WBC is not collected on the stipulated days, with preference given to the preceding day’s results. If more than one WBC is measured on the same day, then the most abnormal results will be recorded.
Secondary outcome measures
-
1.
All-cause mortality at 14, 42, and 90 days.
-
2.
Time to resolution of SIRS (number of days until the patient meets < 2 simultaneous SIRS criteria on a calendar day).
-
3.
Proportion with microbiological relapse (positive blood culture for MSSA or MRSA at least 72 h after a preceding negative culture).
-
4.
Proportion with microbiological treatment failure (positive sterile site culture for MSSA or MRSA at least 14 days after randomisation).
-
5.
Number of surgical procedures performed for the purposes of S. aureus infection source control.
-
6.
Duration of intravenous antibiotic treatment.
-
7.
C. difficile-associated diarrhoea (3 or more loose stools per day along with a positive laboratory test for C. difficile toxin).
-
8.
All cause diarrhoea (3 or more loose stools per day).
-
9.
Slope of CRP curve days 1–14 (i.e. rate of change of CRP over time).
Participant timeline (Table 2)
Eligibility screening
We will identify potential patients as those who meet the two main inclusion criteria of: S. aureus (MSSA and MRSA) identified in a clinically relevant specimen; and clinically defined severe disease (septic shock, necrotising lung/pleural space infection, complicated multifocal skin or soft tissue, or osteoarticular infection). All such identified patients will be screened and assessed for potential enrolment and randomisation. The identification of such participants will typically be through notification by microbiology staff (e.g. when blood cultures are called through and clinical information obtained from the treating clinician) or other medical staff (e.g. infectious diseases, paediatric, ICU). Identification of S. aureus as MSSA or MRSA can be by any acceptable methods, such as nucleic acid amplification of fem/nuc, mec genes, or phenotypic susceptibility methods.
Informed consent
The PI or their delegate will approach an eligible patient or their next of kin (NOK) for a discussion on participation in the trial. Written information will also be provided. An interpreter will be used if necessary. Informed consent from a NOK will only be used in jurisdictions where there is legislative approval to do so, and where the site has research governance approval in place.
Paediatric-specific consent issues
All participants under the age of 18 years will have consent sought from a parent/guardian. Children and adolescents will have the study explained to them using appropriate language. Where deemed appropriate, the adolescent will be asked to co-sign the parent/guardian consent form. If an adolescent does not want to participate in the study, this would be considered a refusal and they will be considered ineligible.
For Aboriginal and Torres Strait Islander children, informed consent can be given by a parent or caregiver who is recognised under Aboriginal customary law or Aboriginal tradition as having decision-making rights over the child’s health care needs and who assumes day-to-day parental responsibilities.
If consenting parents are under the age of 18 years, consent will be sought from the child’s grandparent with a parent signing as a witness.
Randomisation and blinding
Patients will be randomised using a module in the web-based study database. The randomisation list will be generated and held by an independent statistician. Randomisation will be stratified by age (child versus adult) in a 1:1 ratio, in permuted blocks of variable size. Randomisation will not be stratified by site, as an average site is only likely to randomise 5–10 patients and hence the strata will be too small. We will not stratify by MSSA/MRSA status for similar reasons—the strata will be too small, and the study is designed as a pilot study with the aim of assessing for feasibility.
Blinding is not relevant as this will be an open-label trial.
Days 2–90
The PI (or delegate) will visit the patient, review medical documentations, or contact the treating team in person or by phone daily for the first 7 days of randomisation to ensure compliance with the protocol and that the recommended tests are ordered. Case report forms (CRFs) with details on illness severity scores, clinical progress, and blood results will be completed daily. These details can be retrospectively collected up to 72 h after the intended day, allowing for weekends and public holidays. Standard operating procedures will contain step-by-step details on how to recruit patients and collect data.
Endpoint assessment
A panel blinded to treatment allocation will determine the primary endpoint. This will consist of three clinicians, each with expertise in infectious diseases, paediatrics, or critical care. The critical care clinician will chair to overcome discrepancies in the decision-making between the three decision-makers.
The panel will be given an extract of the database which contains all information needed to determine the number of days alive and free of SIRS over the first 14 days but will not be given any information about the use of clindamycin.
If needed, the panel may request further information (e.g. medical record or observation chart extracts with identifying information redacted) for any particular patient.
Discontinuation/withdrawal of participants from trial treatment
Participants or NOK can voluntarily withdraw from study at any time if they wish to. Investigators may also discontinue a participant from the study if deemed appropriate at any time. Participants do not necessarily need to provide explanations for their withdrawal. If withdrawn by the site investigator, reasons for discontinuation will be recorded in CRFs. Participants will not be withdrawn from the study due to suspected adverse reactions of study treatment, unless the participant or their treating clinician requests discontinuation. If the participant or NOK withdraws consent to participate in the study and also withdraws consent for collection of future information, no further evaluations will be performed, and no additional data will be collected. The investigators may retain and continue to use any data collected before such withdrawal of consent. All withdrawal/discontinuation will be discussed with the coordinating investigators.
To avoid missing data, and for use in intention-to-treat analysis, all attempts will be made to collect information at day 90 for participants who discharge early, transfer to another facility, discharge against medical advice, abscond, or are lost to follow-up.
Sample size considerations
Sixty patients will be enrolled, at least 20 of whom are aged < 16 years. As this is a pilot study, the sample size is based on the number achievable, the number needed to determine feasibility, and to refine assumptions and study design.
Data management
Data collection
All initial data collection can either be in paper format (with subsequent entry into the electronic database) or directly entered into the electronic database. For database entry, each variable will have a validation range to minimise entry errors. The coordinating centre will check data for consistency and will seek any missing data with help of the PI.
During randomisation, each participant will be given a unique identifier number. This number and the hospital medical record number will be recorded in each CRF. These two numbers will be cross-checked each time data entry is performed in CRFs to ensure correct documentation. The date, time, and name of the person performing data entry for each entry in the CRF will be mandatory. Any changes made in the CRF will be timed, dated, and signed and the reasons for changes documented. CRFs will not contain participant identifying details such as address, initials, or date of birth, but will contain age at recruitment.
Clinical details will be obtained from medical records (paper and/or electronic), bedside charts, medication charts, pathology results, correspondence notes, and telephone contact with the patient or their NOK. Clarification will be made with the treating team or the participants’ general practitioners if necessary.
Data entry and storage
Data collected in paper CRFs will be entered onto a purpose-built secure web-based database. This will be done at each site by the PI or delegate. Paper CRFs will be securely stored at each site. Data will be retained for at least 10 years after study completion.
Statistical methods
Data reporting will follow CONSORT guidelines for reporting of RCTs.
Continuous variables will be analysed using the Mann–Whitney U test or Student’s t test as appropriate. Proportions will be analysed by Fisher’s exact test or chi-squared tests as appropriate. The 95% confidence interval will be reported for absolute difference in proportions. All-cause mortality will be shown with Kaplan–Meier graphs.
The primary analysis of both primary and secondary endpoints will be according to modified intention-to-treat principles (all participants with data available for the endpoint will be analysed according to the treatment allocation, regardless of what treatment they received).
Secondary per-protocol analysis of all endpoints will be conducted. The per-protocol population is defined as: the combination group that received at least 75% of clindamycin dose; the standard treatment group that received ≤ 1 defined daily dose of clindamycin in the week preceding randomisation; and those with data available for at least 90 days of follow-up.
There will be no planned interim efficacy analyses.
Pre-specified subgroup analyses
The number of subgroup analyses will be limited due to the small sample size:
-
i)
Main treatment was flucloxacillin vs cefazolin (for the MSSA cohort). Recent data suggest better outcomes for SAB with cefazolin versus nafcillin/oxacillin [47], although it is unclear whether this is true, and the effect size is likely to be small.
-
ii)
Those who received > 24 h of lincosamides or other anti-toxin antibiotics (as defined in the eligibility criteria) in the 7 days prior to randomisation, compared with those who did not. Recent receipt of lincosamides is likely to dilute the effect of the randomised intervention.
-
iii)
Children vs adults. The distribution of outcomes is likely to be different in children from that in adults, although the effect of the intervention should not differ.
-
iv)
MSSA vs MRSA infection. MRSA isolates may have higher rates of clindamycin resistance or may involve patients within certain risk groups (e.g. intravenous drug users, haemodialysis patients, or recent hospitalisation).
Data safety and monitoring board
An independent data safety and monitoring board (DSMB) will be established to review the progress of the study and monitor adherence to the protocol, participant recruitment, outcomes, complications, and other issues related to participant safety. They will also monitor the assumptions underlying sample size calculations for the study and alert the investigators if they see substantial departures from the study protocol as the data accumulate.
The DSMB will be composed of experts in infectious diseases (adult representative and paediatric representative) and biostatistics, and an intensivist. The DSMB members will all be independent of the investigators (none of them will be chief investigators or site PIs).
Safety aspects of the trial
Adverse events and adverse reaction (solicited and unsolicited)
Clindamycin is registered in Australia for therapeutic use. Clindamycin has a good safety profile but has some recognised adverse effects [48]. Antibiotic-associated diarrhoea and C. difficile diarrhoea are the most frequently encountered clinical adverse event with clindamycin. Rash, blood dyscrasias, and raised liver enzymes could be difficult to attribute to clindamycin, especially since the trial participants will also be receiving β-lactams and have a severe infection. Nonetheless, these and any other potential adverse events in both standard therapy and combination therapy groups will be monitored and recorded in CRFs. Clindamycin can be ceased if the patient develops C. difficile diarrhoea, severe non-C. difficile diarrhoea, or other serious adverse events.
Vancomycin adverse events include phlebitis, nephrotoxicity, rash, and red-man syndrome (if infused rapidly). Daptomycin adverse events include headache, rash, infection site reactions, increased creatinine kinase, increased liver enzymes, and hypotension. Less common adverse effects of vancomycin include immune-mediated thrombocytopenia and ototoxicity, and those of daptomycin include eosinophilic pneumonia [48].
Adverse events will be graded as follows (Additional file 1: Table S6) [49]:
-
Grade 1: mild symptoms. Causes nil or minimal interference with usual social and functional activities. No intervention indicated.
-
Grade 2: moderate symptoms. Interferes with but does not limit usual social and functional activities. Intervention is indicated.
-
Grade 3: severe symptoms. Inability to perform usual social and functional activities. Intervention or hospitalisation is needed.
-
Grade 4: potentially life-threatening symptoms. Inability to perform basic self-care functions. Intervention is indicated to prevent permanent impairment, persistent disability, or death.
-
Grade 5: death related to adverse event.
Serious adverse events
Serious adverse events (SAEs) are an undesirable experience as a result of the intervention that:
-
◦ results in death;
-
◦ is life-threatening;
-
◦ results in hospitalisation (initial or prolongation);
-
◦ results in disability or permanent damage; or
-
◦ is a medically important event or reaction.
All SAEs that are considered related (possible, probable, or definite) to any of the study drugs (backbone and combination therapy) which met one or more the above definitions require reporting on the SAE form. If the SAE is attributable to disease progression then this does not require expedited reporting in this trial, even when death is the outcome. The site PI is required to report any SAEs that occur at their site to the approving ethics committee in accordance with local guidelines; in addition, the site PI must adhere to any local institutional reporting requirements. Safety reporting will be in line with the National Health and Medical Research Council [50]. Guidance: Safety monitoring and reporting in clinical trials involving therapeutic goods. Canberra: National Health and Medical Research Council.
Suspected unexpected serious adverse reaction
If the SAE is considered unexpected (not listed in approved product information and not attributable to disease progression) and related (possible, probable, or definite) to the investigational drug (clindamycin), it meets the definition of a suspected unexpected serious adverse reaction (SUSAR). All SUSARs must be entered on the SAE CRF and reported to the coordinating investigators (CIs) or delegate within 24 h from the time of the site study team becoming aware of it. If the SUSAR is fatal or life-threatening, the PI will report to the TGA within 7 calendar days of becoming aware and any follow-up reports will be submitted within a further 8 calendar days; all other SUSARs will be reported to the Therapeutic Goods Administration (TGA) within 15 calendar days of the PI becoming aware.
The site PI is required to report any SUSARs that occur at their site to the approving ethics committee in accordance with local guidelines; in addition, the site PI must adhere to any local institutional reporting requirements. Safety reporting will be in line with the NHMRC Guidance: Safety Monitoring and Reporting in Clinical Trials Involving Therapeutic Goods [50].
The relation of adverse event to the treatment will be defined as not related, unlikely, possible, probable, and definite [51].
Ethical considerations
The trial will be conducted in line with the Declaration of Helsinki. Written informed consent will be obtained from all study participants prior to any study procedure. Approval from a lead HREC and site-specific approval will be finalised at each site before the first patient is enrolled at that site. There will be two lead HRECs: one for the NT site (Menzies and NT Department of Health EC00153), and one for the remaining sites as per the National Mutual Acceptance scheme (Hunter New England EC00403).
Dissemination policy
The chief investigators (JSD, SYCT, AB) will have access to the study dataset. The results of this study will be submitted to peer-reviewed journals for publication regardless of the results. They will also be presented at national and/or international scientific meetings. Authorship of the main paper will be all PIs, “on behalf of the CASSETTE study group, the ASID CRN and Australia and New Zealand Paediatric Infectious Diseases Group (ANZPID)”. The CASSETTE Study Group will consist of all CIs and all site PIs, as well as up to two co-investigators per site if relevant.
Discussion
Although animal studies and observational reports support the concept of effective S. aureus exotoxin suppression with clindamycin [17], adequate supporting clinical evidence is lacking. The proposed study is an open-label, pilot, randomised controlled trial to determine whether 7 days of clindamycin in combination with standard therapy will lead to a faster resolution of systemic inflammation than standard therapy alone in adults and children with severe S. aureus infection.
It is unclear whether the anti-toxin effect of clindamycin is retained in S. aureus strains that are resistant to it. This may be a limiting factor for our study design. Fortunately, most of the MSSA isolates in Australia have low rates of constitutive resistance against clindamycin (0.8–2.4% of blood culture isolates) [52,53,54]. Clindamycin resistance is, however, more common in MRSA isolates (16.5–21%) [52, 53]. Inducible resistance to clindamycin due to the presence of a mutation in the erm gene is much more commonly observed as a potential issue in isolates resistant to erythromycin (~ 50% of MRSA isolates from blood culture are erythromycin resistant) [55], although not all erythromycin-resistant strains are resistant to clindamycin. Resistance to macrolides due to efflux pumps does not create resistance to lincosamides (clindamycin and lincomycin). In the peptide tunnel of the 50S ribosome subunit, each of MLSB antibiotics has its own interaction sites. Methylation at the common interaction site at nucleotide A2058/A2059 of 23S rRNA, mediated by an enzyme encoded by one or more erm genes, confers resistance to all MLSB compounds [31, 32]. Mutations at other nucleotides create selective resistance to macrolides, and spares lincosamides [31]. Also, sub-inhibitory concentrations of clindamycin are consistently shown to inhibit S. aureus exotoxins [17]. A recent French study demonstrated that sub-inhibitory concentrations of clindamycin did lead to a dramatic decrease in toxin production in various S. aureus strains with inducible clindamycin resistance (positive D test) [56]. Hence, it is likely that clindamycin retains its efficacy as a toxin blocker in most erm-containing S. aureus strains, with the probable exception of those with constitutive resistance.
In conclusion, the proposed CASSETTE study will assess the effect of clindamycin in patients with severe S. aureus infections and will provide feasibility for a larger RCT. The study will provide further clinical evidence for such a use.
Trial status
This is protocol version 2.0 (18 August 2018). Recruitment started 10 July 2018. Expected completion 31 December 2020.
Availability of data and materials
Not applicable
Abbreviations
- ASID:
-
Australasian Society for Infectious Diseases
- CI:
-
Coordinating investigator
- CONSORT:
-
Consolidated Standards of Reporting Trials
- CRF:
-
Case report form
- CRN:
-
Clinical research network
- CRP:
-
C-reactive protein
- DSMB:
-
Data safety and monitoring board
- FBC:
-
Full blood count
- HITH:
-
Hospital in the home
- HR:
-
Heart rate
- HREC:
-
Human research and ethics committee
- ICU:
-
Intensive care unit
- IV:
-
Intravenous
- Menzies:
-
Menzies School of Health Research Global and Tropical Health Division
- MIC:
-
Minimum inhibitory concentration
- MRSA:
-
Methicillin-resistant Staphylococcus aureus
- MSSA:
-
Methicillin-susceptible Staphylococcus aureus
- NHMRC:
-
National Health and Medical Research Council
- NOK:
-
Next of kin
- PI:
-
Principal Investigator
- PO:
-
Per oral
- PSI:
-
Protein synthesis inhibitor
- PVL:
-
Panton-Valentine leucocidin
- QID:
-
Four times a day
- RCT:
-
Randomised controlled trials
- RR:
-
Respiratory rate
- SAB:
-
Staphylococcus aureus bacteraemia
- SAE:
-
Serious adverse event
- SIRS:
-
Systemic inflammatory response syndrome
- SPIRIT:
-
Standard Protocol Items Recommendations for Interventional Trials
- SSTI:
-
Skin and soft tissue infections
- SUSAR:
-
Suspected unexpected serious adverse reaction
- TDS:
-
Three times a day
- TG:
-
Therapeutic Guidelines (Australia) v15
- TGA:
-
Therapeutic goods administration
- TSST:
-
Toxic shock syndrome toxin
References
Wertheim HFL, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, et al. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis. 2005;5:751–62.
Williams RE. Healthy carriage of Staphylococcus aureus: its prevalence and importance. Bacteriol Rev. 1963;27:56–71.
Wyllie DH, Crook DW, Peto TEA. Mortality after Staphylococcus aureus bacteraemia in two hospitals in Oxfordshire, 1997-2003: cohort study. BMJ. 2006;333:281.
van Hal SJ, Jensen SO, Vaska VL, Espedido BA, Paterson DL, Gosbell IB. Predictors of mortality in Staphylococcus aureus Bacteremia. Clin Microbiol Rev. 2012;25:362–86.
McMullan BJ, Bowen A, Blyth CC, Van Hal S, Korman TM, Buttery J, et al. Epidemiology and mortality of Staphylococcus aureus bacteremia in Australian and New Zealand children. JAMA Pediatr. 2016;170:979–86.
Gillet Y, Issartel B, Vanhems P, Fournet J-C, Lina G, Bes M, et al. Association between Staphylococcus aureus strains carrying gene for Panton-Valentine leukocidin and highly lethal necrotising pneumonia in young immunocompetent patients. Lancet (London, England). 2002;359:753–9.
Gillet Y, Vanhems P, Lina G, Bes M, Vandenesch F, Floret D, et al. Factors predicting mortality in necrotizing community-acquired pneumonia caused by Staphylococcus aureus containing Panton-Valentine leukocidin. Clin Infect Dis. 2007;45:315–21.
Tong SYC, Davis JS, Eichenberger E, Holland TL, Fowler VGJ. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev. 2015;28:603–61.
von Eiff C, Becker K, Machka K, Stammer H, Peters G. Nasal carriage as a source of Staphylococcus aureus bacteremia. Study Group. N Engl J Med. 2001;344:11–6.
Bai AD, Showler A, Burry L, Steinberg M, Ricciuto DR, Fernandes T, et al. Impact of infectious disease consultation on quality of care, mortality, and length of stay in Staphylococcus aureus bacteremia: results from a large multicenter cohort study. Clin Infect Dis. 2015;60:1451–61.
Vogel M, Schmitz RPH, Hagel S, Pletz MW, Gagelmann N, Scherag A, et al. Infectious disease consultation for Staphylococcus aureus bacteremia—a systematic review and meta-analysis. J Infect. 2016;72:19–28.
Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis. 2011;52:285–92.
Thwaites GE, Edgeworth JD, Gkrania-Klotsas E, Kirby A, Tilley R, Torok ME, et al. Clinical management of Staphylococcus aureus bacteraemia. Lancet Infect Dis. 2011;11:208–22.
eTG complete [Internet]. Melbourne: Therapeutic Guidelines Limited; 2015.
Health Protection Agency (UK). Guidance on the diagnosis and management of PVL-associated Staphylococcus aureus infections (PVL-SA) in England. 2nd edition. 2008.
Gillet Y, Dumitrescu O, Tristan A, Dauwalder O, Javouhey E, Floret D, et al. Pragmatic management of Panton-Valentine leukocidin-associated staphylococcal diseases. Int J Antimicrob Agents. 2011;38:457–64.
Campbell AJ, Dotel R, Blyth CC, Davis JS, Tong SYC, Bowen AC. Adjunctive protein synthesis inhibitor antibiotics for toxin suppression in Staphylococcus aureus infections: a systematic appraisal. J Antimicrob Chemother. 2019;74:1–5.
Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–32.
Dinges MM, Orwin PM, Schlievert PM. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev. 2000;13:16–34 table of contents.
Foster TJ. Immune evasion by staphylococci. Nat Rev Microbiol. 2005;3:948–58.
Becker K, Friedrich AW, Lubritz G, Weilert M, Peters G, Von Eiff C. Prevalence of genes encoding pyrogenic toxin superantigens and exfoliative toxins among strains of Staphylococcus aureus isolated from blood and nasal specimens. J Clin Microbiol. 2003;41:1434–9.
Spaulding AR, Salgado-Pabon W, Kohler PL, Horswill AR, Leung DYM, Schlievert PM. Staphylococcal and streptococcal superantigen exotoxins. Clin Microbiol Rev. 2013;26:422–47.
Peacock SJ, Moore CE, Justice A, Kantzanou M, Story L, Mackie K, et al. Virulent combinations of adhesin and toxin genes in natural populations of Staphylococcus aureus. Infect Immun. 2002;70:4987–96.
Hewagama S, Spelman T, Woolley M, McLeod J, Gordon D, Einsiedel L. The epidemiology of Staphylococcus aureus and Panton-Valentine leucocidin (PVL) in Central Australia, 2006-2010. BMC Infect Dis England. 2016;16:382.
Ellington MJ, Hope R, Ganner M, Ganner M, East C, Brick G, et al. Is Panton-Valentine leucocidin associated with the pathogenesis of Staphylococcus aureus bacteraemia in the UK? J Antimicrob Chemother. 2007;60:402–5.
Shallcross LJ, Fragaszy E, Johnson AM, Hayward AC. The role of the Panton-Valentine leucocidin toxin in staphylococcal disease: a systematic review and meta-analysis. Lancet Infect Dis. 2013;13:43–54.
Chua KYL, Monk IR, Lin Y-H, Seemann T, Tuck KL, Porter JL, et al. Hyperexpression of alpha-hemolysin explains enhanced virulence of sequence type 93 community-associated methicillin-resistant Staphylococcus aureus. BMC Microbiol. 2014;14:31.
Tong SYC, Lilliebridge RA, Holt DC, McDonald MI, Currie BJ, Giffard PM. High-resolution melting analysis of the spa locus reveals significant diversity within sequence type 93 methicillin-resistant Staphylococcus aureus from northern Australia. Clin Microbiol Infect. 2009;15:1126–31.
Kong C, Neoh H, Nathan S. Targeting Staphylococcus aureus toxins: a potential form of anti-virulence therapy. Toxins (Basel). 2016;8:72.
Verkaik NJ, Dauwalder O, Antri K, Boubekri I, de Vogel CP, Badiou C, et al. Immunogenicity of toxins during Staphylococcus aureus infection. Clin Infect Dis. 2010;50:61–8.
Poehlsgaard J, Douthwaite S. The bacterial ribosome as a target for antibiotics. Nat Rev Microbiol. 2005;3:870–81.
Wilson DN. The A-Z of bacterial translation inhibitors. Crit Rev Biochem Mol Biol. 2009;44:393–433.
Diep BA, Equils O, Huang DB, Gladue R. Linezolid effects on bacterial toxin production and host immune response: review of the evidence. Curr Ther Res Clin Exp. 2012;73:86–102.
Eagle H. Experimental approach to the problem of treatment failure with penicillin. I. Group A streptococcal infection in mice. Am J Med. 1952;13:389–99.
Stevens DL, Gibbons AE, Bergstrom R, Winn V. The Eagle effect revisited: efficacy of clindamycin, erythromycin, and penicillin in the treatment of streptococcal myositis. J Infect Dis. 1988;158:23–8.
Stevens DL, Ma Y, Salmi DB, McIndoo E, Wallace RJ, Bryant AE. Impact of antibiotics on expression of virulence-associated exotoxin genes in methicillin-sensitive and methicillin-resistant Staphylococcus aureus. J Infect Dis. 2007;195:202–11.
Kernodle DS, McGraw PA, Barg NL, Menzies BE, Voladri RK, Harshman S. Growth of Staphylococcus aureus with nafcillin in vitro induces alpha-toxin production and increases the lethal activity of sterile broth filtrates in a murine model. J Infect Dis. 1995;172:410–9.
Ohlsen K, Ziebuhr W, Koller KP, Hell W, Wichelhaus TA, Hacker J. Effects of subinhibitory concentrations of antibiotics on alpha-toxin (hla) gene expression of methicillin-sensitive and methicillin-resistant Staphylococcus aureus isolates. Antimicrob Agents Chemother. 1998;42:2817–23.
Dumitrescu O, Boisset S, Badiou C, Bes M, Benito Y, Reverdy M-E, et al. Effect of antibiotics on Staphylococcus aureus producing Panton-Valentine leukocidin. Antimicrob Agents Chemother. 2007;51:1515–9.
Dumitrescu O, Badiou C, Bes M, Reverdy M-E, Vandenesch F, Etienne J, et al. Effect of antibiotics, alone and in combination, on Panton-Valentine leukocidin production by a Staphylococcus aureus reference strain. Clin Microbiol Infect. 2008;14:384–8.
Otto MP, Martin E, Badiou C, Lebrun S, Bes M, Vandenesch F, et al. Effects of subinhibitory concentrations of antibiotics on virulence factor expression by community-acquired methicillin-resistant Staphylococcus aureus. J Antimicrob Chemother. 2013;68:1524–32.
Goldstein B, Giroir B, Randolph A. International pediatric sepsis consensus conference: definitions for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care Med. 2005;6:2–8.
Arrieta AC, Bradley JS, Popejoy MW, Bensaci M, Grandhi A, Bokesch P, et al. Randomized multicenter study comparing safety and efficacy of daptomycin versus standard-of-care in pediatric patients with Staphylococcal bacteremia. Pediatr Infect Dis J. 2018;37:893–900.
Children’s Hospital at Westmead. Antimicrobial dosing in renal impairment. 2015. http://www.schn.health.nsw.gov.au/_policies/pdf/2012-8011.pdf. Accessed 6 July 2017.
Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest, vol. 101; 1992. p. 1644–55.
Clinical Excellence Commission, New South Wales Government (Australia). Paediatric Patient Safety Pathways and Guidelines. Available on the webpage: http://www.cec.health.nsw.gov.au/patient-safety-programs/paediatric-patient-safety/pqp-sepsis-kills. Accessed 30 Oct 2018.
McDanel JS, Roghmann M-C, Perencevich EN, Ohl ME, Goto M, Livorsi DJ, et al. Comparative effectiveness of cefazolin versus nafcillin or oxacillin for treatment of methicillin-susceptible Staphylococcus aureus infections complicated by bacteremia: a nationwide cohort study. Clin Infect Dis. 2017;65:100–6.
Australian Medicines Handbook 2017 (online). Adelaide: Australian Medicines Handbook Pty Ltd; 2017. Available from: https://amhonline.amh.net.au.
U.S. Department of Health and Human Services, National Institutes of Health, National Institute of Allergy and Infectious Diseases, Division of AIDS. Division of AIDS (DAIDS) table for grading the severity of adult and pediatric adverse events, corrected version 2.1. 2017.
National Health and Medical Research Council (NHMRC) Guidance: Safety Monitoring and Reporting in Clinical Trials Involving Therapeutic Goods (2016). https://www.nhmrc.gov.au/sites/default/files/images/NHMRC-guidance-safety-monitoring-and-reporting.pdf
Tong SYC, Nelson J, Paterson DL, Fowler VGJ, Howden BP, Cheng AC, et al. CAMERA2—combination antibiotic therapy for methicillin-resistant Staphylococcus aureus infection: study protocol for a randomised controlled trial. Trials. 2016;17:170.
Coombs GW, Nimmo GR, Daly DA, Le TT, Pearson JC, Tan H-L, et al. Australian Staphylococcus aureus Sepsis Outcome Programme annual report, 2013. Commun Dis Intell Q Rep. 2014;38:E309–19.
Coombs GW, Daley DA, Thin Lee Y, Pearson JC, Robinson JO, Nimmo GR, et al. Australian Group on Antimicrobial Resistance Australian Staphylococcus aureus Sepsis Outcome Programme annual report, 2014. Commun Dis Intell Q Rep. 2016;40:E244–54.
The Australian Group on Antimicrobial Resistance (AGAR) 2015 Australian Staphylococcus aureus Sepsis Outcome Program (ASSOP). Staphpath 2017; 2017 25–26 February; Adelaide, Australia; 2017.
Lewis JS 2nd, Jorgensen JH. Inducible clindamycin resistance in Staphylococci: should clinicians and microbiologists be concerned? Clin Infect Dis. 2005;40:280–5.
Hodille E, Badiou C, Bouveyron C, Bes M, Tristan A, Vandenesch F, et al. Clindamycin suppresses virulence expression in inducible clindamycin-resistant Staphylococcus aureus strains. Ann Clin Microbiol Antimicrob. 2018;17:38.
Acknowledgements
The CASSETTE study protocol was presented as an abstract at the International Symposium on Staphylococci and Staphylococcal Infections (ISSSI) 2018, Copenhagen, Denmark. RD received an Australian Postgraduate Awards (APA) Ph.D. scholarship.
Role of the funding body in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript
None
Funding
The study is supported by Australian National Health and Medical Research Council (NHMRC) ID GNT1131932. SYCT is a NHMRC Career Development Fellow (#1145033), AB is a NHMRC Early Career Fellow (#1088735), and JSD is a NHMRC Career Development Fellow (#1083105).
Author information
Authors and Affiliations
Contributions
JSD, SYCT, and AB designed the study. RD performed the literature review and wrote the initial draft of the protocol. JNN, MVNOS, AJC, BJM, PNB, JRF, DPE, OR, and LM all contributed to the revised and final drafts of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Informed consent will be obtained from all study participants.
The following ethics board have approved the study:
-
1.
Hunter New England Human Research Ethics Committee (HNEHREC); Hunter New England Local Health District, New South Wales, Australia (Reference No.: 17/09/20/3.02).
-
2.
Human Research Ethics Committee of the Northern Territory Department of Health and Menzies Scholl of Health Research (Reference No.: 2017-2942).
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Renal dose adjustment, adverse events of interest and their grading, and proposed study sites (DOCX 21 kb)
Additional file 2:
SPIRIT 2013 Checklist: Recommended items to address in a clinical trial protocol and related documents (DOC 124 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dotel, R., Tong, S.Y.C., Bowen, A. et al. CASSETTE—clindamycin adjunctive therapy for severe Staphylococcus aureus treatment evaluation: study protocol for a randomised controlled trial. Trials 20, 353 (2019). https://doi.org/10.1186/s13063-019-3452-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-019-3452-y