
Abstract
Background/rationale
Atrial fibrillation (AF) is frequent and causes great morbidity in the aging population. While initial events may be symptomatic, many patients have silent AF and are at risk of ischemic embolic complications. Timely detection of asymptomatic patients is paramount. The HECTO-AF trial aims to investigate the efficacy of an electrocardiogram (ECG) handheld device for the detection of AF in patients in hospital without a prior diagnosis of AF.
Methods/design
The “Handheld ECG tracking of in-hospital atrial fibrillation” (HECTO-AF) study is a single-center, open-label, randomized controlled trial. The study population consists of all adult patients admitted to a general medicine ward of the University and Hospital of Fribourg throughout the study period. The study will enroll 1600 patients with 1:1 ratio allocation to either the detection group with one-lead handheld ECG recordings twice daily and extra recordings in the case of palpitations, versus a control group undergoing detection of AF as per routine clinical practice. Recordings will be self-performed after dedicated training, and will be independently adjudicated through a specific web-based interface. All enrolled patients will be followed clinically at 1, 2 and 5 years to assess the occurrence of AF, death, non-fatal stroke, systemic embolism, myocardial infarction and bleeding. The primary outcome is incidence of newly detected AF during the hospital stay. Secondary outcomes are incidence of AF, cardiovascular death, stroke, myocardial infarction and bleeding complications at 1, 2 and 5 years.
Discussion
HECTO-AF is an independent randomized study aiming to detect the incidence of silent AF in all-comers hospitalized in general medicine wards.
Trial registration
ClinicalTrials.gov, NCT03197090. Registered on 23 June 2017. Local ethical Committee (CER-VD) registration number: 2017–01594. There are no conflicts of interest to declare.
Similar content being viewed by others

Background
Background and rationale
Atrial fibrillation (AF) is the most common arrhythmia [1, 2], affecting an estimated 3% of the general population above 20 years old [3]. The prevalence has risen over past decades due to shifting demographics resulting in an older population [4]. The prevalence of in-hospital AF is 10–31% [5,6,7]. Patients with AF have increased morbidity and a twofold increase in mortality when compared with the general population [8], and it is estimated that one third of patients with AF will be hospitalized at least once a year due to worsening heart failure, cardioembolic events or progressive dementia [9]. Thus, AF negatively impacts public health.
When symptomatic, AF may induce palpitations, chest pain, fatigue, and exercise intolerance. However, at least a third of patients with AF are asymptomatic. Furthermore, only 20% of symptomatic patients with AF will have symptoms temporally related to their AF episodes [10]. For this reason, the prevalence of AF may be much higher than has been previously reported. The EMBRACE [11] and CRYSTAL-AF trials [12] demonstrated that prolonged electrocardiogram (ECG) monitoring significantly improved detection of AF in patients with cryptogenic stroke or transient ischemic attack (TIA). Recognizing silent AF in the general population, and subsequent treatment could reduce morbidity and mortality [13].
The European Society of Cardiology (ESC) guidelines recommend AF screening by palpation or resting ECG in all patients > 65 years of age, and especially in populations at increased risk [14]. Recently, single-lead ECG recording devices have been developed, allowing quick, precise and non-invasive detection of AF.
The present study aims to determine the incidence of silent AF using a single-lead ECG handheld device in patients hospitalized in the department of general internal medicine of the University and Hospital of Fribourg, Switzerland. Participants will be randomized to either the intervention screening group with twice-daily single-lead ECG recordings and 12-lead ECG in the case of symptoms or to the comparator control group consisting of standard care.
Methods
Participants, interventions, and outcomes
Study setting
This is a single-center, open-label, randomized controlled trial with an allocation ratio of 1:1 conducted in the University and Hospital of Fribourg, Switzerland. All adult patients admitted to the General Internal Medicine Department will be screened for study enrollment. The Standard Protocol Items: Recommendation for Interventional Trials (SPIRIT) checklist can be found in Additional file 1 and the SPIRIT figure is included in the main body of the manuscript (Fig. 1).

Study flowchart. Participants are randomized 1:1 to either handheld single-lead electrocardiogram (ECG) (Zenicor) screening or the control group
Eligibility criteria
All patients 18 years or older admitted to the General Internal Medicine Ward are eligible. Patients with known or previously documented AF, patients with cardiac pacemakers or implantable cardioverter-defibrillators and those unable or unwilling to provide written informed consent will be excluded.
Interventions
Patients allocated to the intervention group will undergo single-lead handheld ECG recordings using the Zenicor device (https://zenicor.com/), twice daily and extra recordings in the case of palpitations during the period of hospitalization. Recordings will be self-performed after dedicated training and according to the Zenicor user manual.
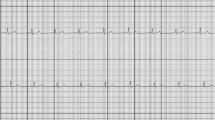
The thumbs of the patient are placed onto the device during 30 s to yield a single-lead ECG recording, which is stored and subsequently transmitted to a central server for clinical analysis. The device has been shown to have higher sensitivity for detection of AF than the conventional 24-h Holter device (sensitivity (SN) 96%, specificity (SP) 92%) [15]. The Zenicor ECG device was chosen for ease of use and reliability, after a test phase in our outpatient clinic including two other devices: the KardiaMobile (https://www.alivecor.com/) and the ME90 mobile ECG (https://www.beurer.com/web/gb/products/medical/ecg-and-pulse-oximeter/mobile-ecg-device/). Examples of recordings are depicted in Fig. 2. The trial follows the instruction of the European Directive on medical devices 93/42/EEC and the ISO Norm 14,155 and ISO 14971, the Swiss Law and Swiss regulatory authority requirements. A specially trained nurse will check for appropriate recording after measurement of routine vital parameters during daily rounds.

Electrocardiogram (ECG) recording with Zenicor, showing atrial fibrillation (AF) (a) and sinus rhythm (b)
All recordings will be independently adjudicated through a specific Web-based interface. Patients allocated to the control group will undergo detection as per routine clinical practice (i.e. symptom-based, in the case of suspicion on auscultation or during measurement of vital parameters if prescribed). The only foreseeable discontinuation criterion in either group would be participant request. Compliance in the intervention group is ensured by the presence of a dedicated nurse and twice-daily Web-based checks for ECG compliance by study investigators.
Patients with new onset of AF
Each patient with newly diagnosed AF will be treated according to the 2016 ESC guidelines on the management of AF (i.e. on anticoagulation, rhythm and/or rate control therapy). Additional workup including 12-lead ECG, laboratory investigations and further cardiological workup such as echocardiography, will be organized as per guidelines.
Outcomes
The primary outcome is the incidence of new-onset AF defined as a 30-s recording of irregular rhythm without p waves during hospitalization [14]. Two independent cardiologists will examine each case of newly diagnosed AF using the Zenicor device. In the case of uncertainty, additional recordings such as 12-lead ECG, 24-h Holter recording or 7-day R-Test may be required.
The secondary outcomes are the incidence of ischemic stroke, systemic embolism, cardiovascular death and myocardial infarction during hospitalization and at 1, 2 and 5 years. Secondary endpoints will be assessed by a telephone interview. If the patient is unavailable, the data will be retrieved from the referring physician/general practitioner, the hospital database or family/relatives of the patient. Local cantonal death registries will be consulted if the information is otherwise unavailable.
Participant timeline
The timeline for enrollment, interventions, and investigations is outlined in Fig. 3.

Timetable of investigations, interventions and assessments. ECG, electrocardiogram; FU, follow up; y, year. Primary outcome: new-onset atrial fibrillation (AF). Secondary outcome: incidence of ischemic stroke or systemic embolism, myocardial infarction, cardiovascular death or heart failure
Sample size
Based on previous research, we estimate to detect AF in 1% of patients in the control group and in 3% of patients in the intervention group. The inclusion of 745 patients per group (total 1490) would yield power of 80% to detect this 2% difference between groups. We seek to include 800 patients per group (total 1600) to account for ineffective measurements, post-inclusion dropouts or other unforeseen dropouts.
Recruitment
Recruitment will be carried out by one full-time study nurse. All patients admitted to the General Internal Medicine Department at the Hospital of Fribourg will be screened. If the eligibility criteria are met the patient will be formally enrolled. All other nurses and physicians in the department have been informed of the study and trained in the use of handheld ECG devices.
Assignment of interventions
Allocation
The randomization sequence was generated via computer (randomizer.org). Patients are randomly assigned in a 1:1 ratio to single-lead Zenicor ECG recording or standard care. The randomization list will be sealed in opaque envelopes and kept by a secretary not involved in the study to guarantee allocation blinding.
Blinding
There is no blinding or masking in the study. The statistician is the only individual blinded to treatment allocation.
Data collection, management and analysis
Data collection methods
The following baseline characteristics will be collected by the study nurse from the patient’s medical records: patient demographics (age, gender, body mass index) and concomitant diseases including but not limited to hypertension, dyslipidemia, diabetes mellitus, heart failure, vascular disease, previous stroke/thromboembolism, renal failure, chronic respiratory disease and other endocrine anomalies (e.g. thyroid disease). Data on other variables will be collected, including symptoms such as palpitations, chest pain, the CHA2DS2-VASc clinical prediction score for risk of stroke and the HAS-BLED score for risk of major bleeding, diagnosis for index hospitalization and medical treatment. Characteristics of screened patients excluded from or unwilling to participate in the trial will not be collected. Selection bias may arise because these patients might differ from the study population with regard to the aforementioned characteristics, which in turn would represent a major study limitation.
Data management
Patient characteristics, study outcomes and clinical follow up will be recorded in an electronic case report form (eCRF; REDcap Software, Vanderbilt University, Nashville, TN, USA [16]). A trained study nurse will complete data management procedures, including coding (through de-identified numbers), verification, validation, security and storage of the database. The investigators will have access to the protocol, dataset and statistical code during and after the study for publication and dissemination. The study nurse will only have access to the dataset during the study time. During the clinical trial, data will be accurately recorded and the original documents will be stored at the clinical trials unit of the University and Hospital of Fribourg, under locked conditions when not in use. At the end of the study, Zenicor ECG recordings will be saved in PDF form and stored for 10 years. The server is in an ISO27001-certified secure data center. The data collected will not include identifiable references to the subjects. The confidentiality of records will be protected.
Statistical methods
We plan to enroll 800 patients in each group. Sample size calculations were performed using STATA (StataCorp. 2013. Stata Statistical Software: Release 13. College Station, TX, USA: StataCorp LP). Based on previous figures, the expected annual detection rate is 3% in the Zenicor group and 1% in the control group. The inclusion of 745 patients per group would yield power of 80% at a two-tailed significance level of α = 0.05. To account for ineffective measurements, post-inclusion dropouts or other unforeseen dropouts, we seek to include 800 patients per group. Categorical variables will be reported as counts and percentages. Continuous variables are presented as mean ± standard deviation or median (25–75%) interquartile range according to their distribution. Normality will be assessed by visual inspection of histograms, the computation of Q-Q plots and the Shapiro-Wilk test. Categorical variables will be compared using the chi-square or Fisher’s exact test as appropriate. Continuous variables will be analyzed using Student’s t test or the Wilcoxon rank-sum test according to the data distribution. Survival free from the occurrence of the clinical end points will be compared using the log-rank test and plotted as Kaplan-Meier survival functions. All statistical analyses will be performed using dedicated software (Stata version 15, StataCorp LP, College Station, TX, USA: StataCorp LP) at a two-tailed significance level of α = 0.05. The null hypothesis to be rejected is that there is no difference in the percentage of patients with newly detected AF in the control and the intervention group. The primary analysis pertains to the proportion patients with newly detected AF. Univariate methods (chi-square test) will be employed. If a significant difference in baseline characteristics is observed, then logistic regression will be performed to correct for baseline imbalances.
A secondary analysis will focus on subgroups. Pre-specified subgroups comprise gender, age, diabetes mellitus, hypertension, ischemic heart disease, congestive heart failure and all subcategories of the CHA2DS2-VASc score. Additional analysis will be performed on clinical follow up. Event-free survival in the intervention and the control group will be compared at 1, 2 and 5 years using the Kaplan-Meier method. Adjustment for imbalances will be performed using Cox proportional hazards given that the data do not violate the proportionality assumption.
Missing data will be omitted pairwise from the analyses. Whenever data are missing, this will be explicitly stated in the final results of the study. Missing data arising from loss to follow up will be censored. The number of dropouts will be provided in a flow-chart along with the reasons for the dropouts.
Monitoring
This is a monitoring study with no additional risks to the patients so no external data monitoring committee is required. The principal investigator and a review board composed of the statistician and the study nurse will perform ongoing monitoring. An interim analysis is planned after inclusion of the first 800 patients for quality management; there will be no early discontinuation of the trial.
Harms
No harm is to be expected from the intervention itself. However, active screening for AF can lead to both false positive or false negative results. This may lead to unwarranted anticoagulant treatment, exposing the patient to an increased risk of bleeding. Conversely, in misdiagnosed patients it may give a false sense of security and increased risk of stroke in patients with high CHA2DS2-VASc scores. To minimize this, as previously mentioned, two independent cardiologists will review every case of new AF diagnosed by the Zenicor device. In the case of uncertainty, additional recordings, e.g. 12-lead ECG, 24-h Holter ECG or 7-day R-Test may be performed.
Ethics and dissemination
Research ethics approval
This study is conducted in compliance with the current version of the Declaration of Helsinki. The research project was approved by the local ethics committee of Vaud, Switzerland (CER-VD 2017–01594).
Protocol amendments
The principal investigator is responsible for communicating important protocol modifications to the ethics committee and to the competent authorities including the clinical trial registry (ClinicalTrials.gov).
Consent
The study nurse will collect patient information at day 1 and provide a consent form with details on trial rationale, interventions, potential benefits and harms. The patients will be given up to 24 h to consider participation. Once having provided written informed consent, the patient will be enrolled and randomized to one of the treatment groups. Patients can withdraw their consent unconditionally, at any time and without any justification. Medical data that have been collected to date will, however, be analyzed.
Confidentiality
The investigators will comply with local privacy laws. Anonymity of the participants will be guaranteed when presenting the data at scientific meetings or publishing them in scientific journals. Individual subject medical information obtained as a result of this study is considered confidential and disclosure to third parties is prohibited.
Access to data
Data will be stored physically and electronically on the personal computers at the clinical trials unit at the University and Hospital of Fribourg. Physical data are protected by restricted access to their location. Electronic data are protected by the IT-Services of the state of Fribourg (SITEL services). The investigators will have access to the protocol, dataset, and statistical code during and after the study for publication and dissemination. The study nurse will only have access to the dataset during the study period.
Declaration of interest
The trial is supported by an unrestricted grant from the Fonds Scientifique Cardiovasculaire Fribourg.
Dissemination policy
The study results will be disseminated within the department of General Internal Medicine, and are intended to be published in peer-reviewed medical journals and communicated at medical conferences.
Discussion
Silent AF is common [17], especially in older patients and those with heart failure [18], and may lead to stroke or death [19, 20]. The 2016 ESC guidelines encourage systematic AF screening programs in at-risk populations [14], which include most patients admitted to general medicine wards, who are older and have several comorbidities. It has been shown that the CHADS2 and CHA2DS2-VASc scores are directly associated with the incidence of new-onset AF and reliably predict AF [21]. A recent study assessing the performance of handheld ECG devices to detect AF in cardiology and geriatric wards [22] found acceptable sensitivity (81–89%) and specificity (94–97%) compared to standard ECG.
The Zenicor ECG is a well-validated device that is non-invasive and easy to use. The product has been used regularly since 2010 in over 350 clinics in Europe. Sensitivity and specificity compared to 12-lead ECG are 96% and 92%, respectively [23]. The device has been evaluated for AF screening and has shown higher sensitivity for detection of silent AF compared to conventional 24-h Holter recordings [24, 25]. One of the advantages compared to other devices includes direct mobile-network transmission to a Web-based central database for rapid online analysis.
A recent Cochrane review on systematic AF screening suggests that future studies should examine the effectiveness of alternative screening strategies such as handheld devices [26]. As previously mentioned, timely detection of silent AF could have major benefits to patients by decreasing both morbidity and mortality. Whether or not this is cost-effective and acceptable to a healthcare network still needs to be determined. The HECTO-AF trial will assess the use of a handheld single-lead ECG device for the screening of silent AF in patients admitted to a general medicine ward.
Trial status
Recruitment started on 1 March 2018; the planned completion date is 30 March 2019.
Abbreviations
- AF:
-
Atrial fibrillation
- CER-VD:
-
Ethics committee of Vaud
- CRYSTAL-AF:
-
Cryptogenic stroke and underlying atrial fibrillation
- ECG:
-
Electrocardiogram
- EMBRACE:
-
30-Day cardiac event monitor belt for recording atrial fibrillation after a cerebral ischemic event
- ESC:
-
European Society of Cardiology
- HECTO-AF:
-
Handheld ECG tracking of in-hospital atrial fibrillation
- R-test:
-
Electrocardiogram event recorder
- SITEL:
-
Services of the state of Fribourg
- SN:
-
Sensitivity
- SP:
-
Specificity
- TIA:
-
Transient ischemic attack
References
Friberg L, Bergfeldt L. Atrial fibrillation prevalence revisited. J Intern Med. 2013;274(5):461–8.
Heeringa J, van der Kuip DAM, Hofman A, Kors JA, van Herpen G, Stricker BHC, et al. Prevalence, incidence and lifetime risk of atrial fibrillation: the Rotterdam study. Eur Heart J. 2006;27(8):949–53.
Björck S, Palaszewski B, Friberg L, Bergfeldt L. Atrial fibrillation, stroke risk, and warfarin therapy revisited: a population-based study. Stroke. 2013;44(11):3103–8.
Krahn AD, Manfreda J, Tate RB, Mathewson FA, Cuddy TE. The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba follow-up study. Am J Med. 1995;98(5):476–84.
Stewart FM, Singh Y, Persson S, Gamble GD, Braatvedt GD. Atrial fibrillation: prevalence and management in an acute general medical unit. Aust NZ J Med. 1999;29(1):51–8.
López Soto A, Formiga F, Bosch X, García Alegría J. En representación de los investigadores del estudio ESFINGE. [Prevalence of atrial fibrillation and related factors in hospitalized old patients: ESFINGE study]. Med Clin (Barc). 2012;138(6):231–7.
Campanini M, Frediani R, Artom A, Pinna G, Valerio A, La Regina M, et al. Real-world management of atrial fibrillation in internal medicine units: the FADOI “FALP” observational study. J Cardiovasc Med Hagerstown Md. 2013;14(1):26–34.
Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham study. Stroke. 1991;22(8):983–8.
Kim MH, Johnston SS, Chu B-C, Dalal MR, Schulman KL. Estimation of total incremental health care costs in patients with atrial fibrillation in the United States. Circ Cardiovasc Qual Outcomes. 2011;4(3):313–20.
Keach JW, Bradley SM, Turakhia MP, Maddox TM. Early detection of occult atrial fibrillation and stroke prevention. Heart. 2015;101(7):1097–102.
Gladstone DJ, Spring M, Dorian P, Panzov V, Thorpe KE, Hall J, et al. Atrial fibrillation in patients with cryptogenic stroke. N Engl J Med. 2014;370(26):2467–77.
Sanna T, Diener HC, Passman RS, Di Lazzaro V, Bernstein RA, Morillo CA, et al. Cryptogenic stroke and underlying atrial fibrillation. N Engl J Med. 2014;370(26):2478–86.
Svennberg E, Engdahl J, Al-Khalili F, Friberg L, Frykman V, Rosenqvist M. Mass screening for untreated atrial fibrillation: the STROKESTOP study. Circulation. 2015;131(25):2176–84.
Kirchhof P, Benussi S, Kotecha D, Ahlsson A, Atar D, Casadei B, for the ESC Scientific Document Group, et al. 2016 ESC guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016;37(38):2893–962.
Hendrikx T, Rosenqvist M, Wester P, Sandström H, Hörnsten R. Intermittent short ECG recording is more effective than 24-hour Holter ECG in detection of arrhythmias. BMC Cardiovasc Disorders. 2014;14:41. https://doi.org/10.1186/1471-2261-14-41.
Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)–a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–81.
Savelieva I, Camm AJ. Clinical relevance of silent atrial fibrillation: prevalence, prognosis, quality of life, and management. J Interv Card Electrophysiol. 2000;4:369–82.
Davis RC, Hobbs FD, Kenkre JE, Roalfe AK, Iles R, Lip GY, Davies MK. Prevalence of atrial fibrillation in the general population and in high-risk groups: the ECHOES study. Europace. 2012;14:1553–9.
Steinberg BA, Hellkamp AS, Lokhnygina Y, Patel MR, Breithardt G, Hankey GJ, et al. Higher risk of death and stroke in patients with persistent vs. paroxysmal atrial fibrillation: results from the ROCKET-AF trial. Eur Heart J. 2015;36:288–96.
Vanassche T, Lauw MN, Eikelboom JW, Healey JS, Hart RG, Alings M, et al. Risk of ischaemic stroke according to pattern of atrial fibrillation: analysis of 6563 aspirin-treated patients in ACTIVE-A and AVERROES. Eur Heart J. 2015;36:281–287a.
Saliba W, Gronich N, Barnett-Griness O, Rennert G. Usefulness of CHADS2 and CHA2DS2-VASc scores in the prediction of new-onset atrial fibrillation: a population-based study. Am J Med. 2016;129(8):843–9.
Desteghe L, Raymaekers Z, Lutin M, Vijgen J, Dilling-Boer D, Koopman P, et al. Performance of handheld electrocardiogram devices to detect atrial fibrillation in a cardiology and geriatric ward setting. Europace. 2017;19(1):29–39.
Doliwa PS, Frykman V, Rosenqvist M. Short-term ECG for out of hospital detection of silent atrial fibrillation episodes. Scand Cardiovasc J. 2009;43(3):163–8.
Doliwa PS, Rosenqvist M, Frykman V. Paroxysmal atrial fibrillation with silent episodes: intermittent versus continuous monitoring. Scand Cardiovasc. 2012;46(3):144–8.
Doliwa Sobocinski P, Anggardh Rooth E, Frykman Kull V, von Arbin M, Wallen H, Rosenqvist M. Improved screening for silent atrial fibrillation after ischaemic stroke. Europace. 2012;14(8):1112–6.
Moran PS, Teljeur C, Ryan M, Smith SM. Systematic screening for the detection of atrial fibrillation. Cochrane Database Syst Rev. 2016;6:CD009586.
Acknowledgements
The authors would like to thank the nursing staff in the Department of Internal Medicine, University and Hospital of Fribourg, for accommodating and assisting with the conduct of this trial.
Funding
The trial is supported by an unrestricted grant from Fond Scientifique Cardiovasculaire Fribourg. The manufacturer of the Zenicor devices is not involved in the study design, data collection, data analysis, data interpretation or writing of the manuscript. No financial support is received from the manufacturer.
Availability of data and materials
Not applicable to this article.
Author information
Authors and Affiliations
Contributions
MM, Department of Internal Medicine, University and Hospital of Fribourg, Switzerland, contributed to study design, data management and analysis and created the first draft of the manuscript. SS, Department of Internal Medicine, University and Hospital of Fribourg, Switzerland, contributed to study design, data management and analysis and the draft of the manuscript. DH, Department of Internal Medicine, University and Hospital of Fribourg, Switzerland, contributed to the design of the study. YF, Department of Cardiology, University and Hospital of Fribourg, Switzerland, contributed to data collection and analysis. SC, Department of Cardiology, University and Hospital of Fribourg, Switzerland, contributed to study design, data management and analysis. DA and SP, Department of Cardiology, University and Hospital of Fribourg, Switzerland, are co-senior authors, contributed to study design, data management and analysis and drafted the manuscript. All authors revised the manuscript critically for important intellectual content and consented to the current publication. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the local ethics committee of Vaud (Switzerland, CER-VD 2017–01594).
Consent for publication
All authors consented to publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional information
Drs Schukraft and Mancinetti are co first authors.
Additional file
Additional file 1:
SPIRIT 2013 checklist. (DOC 121 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Schukraft, S., Mancinetti, M., Hayoz, D. et al. Handheld ECG Tracking of in-hOspital Atrial Fibrillation The HECTO-AF trial Clinical Study Protocol. Trials 20, 92 (2019). https://doi.org/10.1186/s13063-019-3189-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-019-3189-7