
Abstract
Background
Thermal ablation techniques have become the first-line treatment of truncal veins in the management of chronic venous disease (CVD). Despite excellent outcomes, these methods are often associated with pain; generally due to their use of heat and the necessity of fluid infiltration around the vein. More recently, novel non-thermal techniques, such as mechanochemical ablation (MOCA) and cyanoacrylate adhesive (CAE) have been developed to overcome these unwelcome effects. So far, the novel techniques have been found to have similar efficacy to thermal methods, yet no direct comparisons between the non-thermal treatment techniques have been conducted to date, giving rise to this study.
Methods/design
This is a prospective, multicentre, randomised clinical trial, recruiting patients with truncal saphenous incompetence. Patients will be randomised to undergo either MOCA or CAE truncal ablation, followed by treatment of any varicosities. All patients will be required to wear compression stockings for 4 days post intervention. The primary outcome measure is the pain score immediately following completion of truncal ablation, measured by a 100-mm Visual Analogue Scale (VAS). The secondary outcomes are entire treatment pain scores, clinical scores, quality of life scores, occlusion rates, time to return to usual activities/work at 2 weeks, 3, 6 and 12 months. Re-intervention rate will be considered from the third month. Cost-effectiveness will be assessed for each intervention at 12 months. The study is powered to detect a mean 10-mm difference in maximum pain score. Allowing for loss to follow-up, the total target recruitment is 180 patients.
Discussion
The study will be the first study to compare MOCA against CAE and is designed to determine which method causes less pain. Completion of this study is expected to be the end of 2019.
Trial registration
ClinicalTrials.gov, ID: NCT03392753. Registered on 17 November 2017.
Similar content being viewed by others


Background
Varicose veins are common and are known to affect approximately one third of the population [1]. Chronic venous disease (CVD) has been demonstrated to have a negative impact on the quality of life (QoL) of patients and treatment of varicose veins has been demonstrated to lead to improvement in the QoL of patients [2,3,4]. Over the past decade, new endovenous techniques have been introduced, and these are felt to be cost-effective, especially when performed in an outpatient or ‘office-based’ setting [5]. These endovenous ablation methods (almost exclusively radiofrequency ablation (RFA) or endovenous laser ablation (EVLA)) employ mainly thermal energy to treat varicose veins. The American Venous Forum (AVF) and the National Institute of Clinical Excellence (NICE) guidelines published in July 2013 recommended the use of endovenous thermal ablation techniques, namely radiofrequency ablation (RFA) or endovenous laser ablation (EVLA), as first-line treatment for truncal reflux [6,7,8]. Occlusion rates of greater than 90% have been demonstrated in studies looking at these two methods at up to 5 years of follow-up [9,10,11,12,13,14].
However, because these methods make use of thermal energy to denature the venous wall, they have the potential to cause pain, skin burns, skin pigmentation, nerve damage and even arteriovenous fistula formation [15, 16]. To minimise these possible complications and to allow effective treatment, tumescent anaesthesia must be infiltrated around the vein to be treated. This, in turn, can be a source of discomfort to patients.
More recently, newer non-thermal, non-tumescent ablation techniques (NTNTs) have been introduced in an attempt to reduce these complications. Mechanochemical ablation (MOCA) and cyanoacrylate adhesive injection (CAE) are two examples of these NTNTs [17, 18]. So far, pain has been shown to be less than [19], or comparable to [18], thermal methods, but also equivalent to them in terms of QoL improvement, time to return to normal activities and occlusion rates [18, 19]. This indicates that they may, one day, be considered favourable to endothermal ablation.
NICE has also recently produced interventional procedure guidance for the use of both MOCA [20] and CAE [21].
To date, there has not been a direct head-to-head comparison of these two non-thermal methods. We, propose to undertake a randomised controlled study comparing MOCA and CAE in the treatment of varicose veins.
Objectives
The main aim of this study is to detect which method causes less pain. Secondary objectives are to assess the pain level over the ensuing few days, change in QoL, clinical severity score, degree of bruising and inflammation, time to return to normal activities/work, re-intervention rates as well as the cost-effectiveness of each method.
Trial design
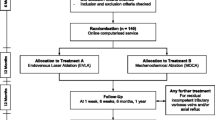
This is a prospective, multicentre, randomised clinical trial comparing MOCA to CAE in the treatment of truncal saphenous incompetence. It follows the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines shown in the SPIRIT Checklist and Figure in Additional file 1 and Fig. 1, respectively. Patients are randomised into group A (MOCA) or group B (CAE). Only the endovenous ablation part of the procedure will be randomised, while the decision as to whether patients should receive treatment of their varicose tributaries will be at the discretion of the clinical team.

Schedule of enrolment, interventions and assessments according to the SPIRIT 2013 Statement: Defining Standard Protocol Items for Clinical Trials. CEAP Clinical Etiology Anatomy Pathophysiology, VCSS Venous Clinical Severity Score, EQ-VAS EuroQol’s Visual Analogue Scale, AVVQ Aberdeen Varicose Vein Questionnaire, CIVQ-14 Chronic Venous Insufficiency Questionnaire, EQ-5D EuroQol’s 5-domain Utility Index, Pain Score VAS Visual Analogue Scale, DUS Duplex ultrasound, MOCA mechanochemical ablation, CAE cyanoacrylate adhesive
Methods/design
Study settings
This study is conducted at Charing Cross Hospital (Imperial College London, UK), Singapore General Hospital and Sengkang General Hospital (Singapore).
Eligibility criteria
Patients who have primary great saphenous (GSV) or short saphenous vein (SSV) vein reflux > 0.5 s on Duplex ultrasound (DUS) scanning and who are aged over 18 years will be included. Exclusion criteria are: current deep vein thrombosis; recurrent varicose veins; arterial disease (ABPI < 0.8); venous diameter < 3 mm; patients who are unwilling to participate; inability or unwillingness to complete questionnaires; adverse reaction to sclerosant or cyanoacrylate or involvement in another venous trial in the past 6 months.
Interventions
All interventions are performed under ultrasound guidance and local anaesthesia and are carried out by vascular surgeons who are experienced in both methods.
For both methods, the truncal vein (GSV or SSV) is cannulated under ultrasound guidance using the Seldinger technique after injection of local anaesthetic (1% lidocaine). The catheter-wire tip is placed 5 cm for the CAE group or 2 cm for the MOCA group distal to the sapheno-femoral junction (SFJ) or sapheno-popliteal junction (SPJ). Cannulation is obtained at the most distal point of venous reflux.
All patients are treated with the ClariVein® mechanochemical ablation device (Vascular Insights, Madison, CT, USA), or the VenaSeal™ Closure System (Medtronic, Minneapolis, MN, USA). The methods used for MOCA and for CAE are described in detail by van Eekeren et al. [22] and by Almeida et al. [23], respectively.
Following all procedures, an ultrasound scan is performed to assess both the treated truncal vein and the deep veins. Concomitant phlebectomy or foam sclerotherapy will then be carried out if deemed necessary and clinically indicated by the clinical team.
The duration of the procedure is defined as from the time of insertion of the cannula into the vein to the time of removal the catheter. The total volume of liquid sclerosant, the amount of proprietary cyanoacrylate glue and the length of the treated vein are recorded.
Re-intervention of the treated saphenous veins will not be considered until at least 3 months after the initial procedure.
Primary outcome
The primary outcome measure is to record the pain score immediately following completion of the endovenous ablation using a 100-mm Visual Analogue Scale (VAS) [19, 22, 24].
Secondary outcome
The secondary outcomes are to compare the two treatment groups with respect to:
-
The pain score at the end of the procedure (including tributary treatment)
-
QoL scores at baseline, 2 weeks, 3 months, 6 months and 12 months using the EuroQol 5-domain Utility Index (EQ-5D), the Aberdeen Varicose Vein Questionnaire (AVVQ) and the Chronic Venous Insufficiency Questionnaire (CIVIQ-14) scores
-
Clinical change using the Venous Clinical Severity Score (VCSS) at baseline, 2 weeks, 3 months, 6 months and 12 months
-
The pain score over the first 10 days, recorded as a number on a scale of 0–10 (0 means no pain, 10 means worst imaginable pain) [25]
-
The degree of bruising at 2 weeks using an ecchymosis score with a 5-point scale [26]
-
The time taken to return to work and normal activities
-
Occlusion rates at 3 months, 6 months and 12 months
-
Re-intervention rate at 12 months
-
Comparison of the cost-effectiveness of each intervention at 12 months
Sample size and study duration
The study is designed to assess for equivalence. Based on previous studies, 10 mm is a clinically appropriate minimum important difference [19, 22, 24]. We estimated the sample size needed to observe a mean difference of at least 10 mm (standard deviation: 20 mm) between the two interventions. With power set at 80% and 5% significance equivalence, we need to recruit 128 patients (64 patients per group). If a dropout rate from follow-up of 30% is estimated, the total number to recruit is 183. If at least three patients are recruited per week, 156 patients could potentially be randomised over 1 year, and 180 patients over 60 weeks (approximately 14 months). Thus, with 12-month follow-up the study will run for 26 months with a target recruitment of 180 patients.
Recruitment
Patients referred for treatment of symptomatic varicose veins are recruited if they are found to have primary GSV or SSV incompetence on the DUS. Patients are identified in clinic and provided with further information regarding the study.
On the day of their procedures, patients are asked to sign a study consent form for inclusion in the study. They will then be allocated a study number. Patients are randomised to receive MOCA (group A) or CAE (group B) to treat their saphenous veins (Fig. 2).

Trial flowchart
At baseline, basic demographic data is collected from each patient. Patients are also be asked to provide their contact details to enable correspondence for follow-up appointments.
Patients are asked to complete QoL questionnaires using the EQ-5D, AVVQ and the CIVIQ-14, and have their clinical scores assessed using Clinical Etiological Anatomical and Pathophysiological (CEAP) classification and VCSS.
Assessment of pain
Immediately following completion of the endovenous ablation, patients are requested to record their pain score using a 100-mm VAS. Once the pain score has been recorded, concomitant phlebectomy or foam sclerotherapy will then be carried out if deemed appropriate by the clinical team. At the end of this part of the procedure, the pain score is again assessed, using the VAS and number scale.
Post procedure
Patients are allowed to leave hospital once they have spent a complication-free period in the department. On discharge, all patients are provided with compression stockings (class II 18–24 mmHg) to wear for 4 days. They are also being provided with a diary to record their post-procedural pain every day for the first 10 post-operative days using a validated VAS and to record when they return to their normal activities and are back to work. They are encouraged to mobilise as much as possible and to resume their normal activities when they feel capable of doing so.
Follow-up treatment periods
Patients are requested to attend a follow-up for research purposes at 2 weeks, 3 months, 6 and 12 months (Fig. 1). The patient’s general practitioner will also be sent a letter to inform them of their patient’s participation in the study, with the consent of the patient.
Follow-up at 2 weeks
At the 2-week follow-up, the diary containing details of the pain scores and how soon patients were able to return to normal activities/work are collected. In addition, patients are asked about, and examined for, any bruising (using an ecchymosis score with a 5-point scale) [26] or phlebitis that followed their procedure. They will undergo a clinical examination and the VCSS will be recorded and will be asked to fill in the EQ-5D, AVVQ and CIVIQ-14 scores. No decision regarding re-treatment will be taken at this point.
Follow-up at 3 months, 6 months and 12 months
At the 3-month, 6-month and 12-month follow-ups, patients undergo further clinical examination, and their VCSS is recorded. They are asked to complete the EQ-5D, AVVQ and the CIVIQ-14 scores. They also undergo further venous DUS scanning to confirm occlusion of the treated vein.
From the third month following treatment, patients found to have recannulation of their treated truncal veins will be assessed to establish if they are symptomatic and require re-intervention. The method used for re-intervention will be dependent on the choice of the consultant in charge of the patient.
Randomisation
On the day of treatment, written consent forms are obtained. Patients are then allocated randomly to one of the two intervention groups by equal randomisation using an online computerised randomisation system (SealedEnvelope, London, UK).
Blinding
Blinding after randomisation to interventions is not possible from either the participant or the clinical team because of nature of the interventions.
Following the intervention, clinician review and DUS will be performed independently by assessors who are blinded to the group allocation.
Data collection and confidentiality
All patient data will be anonymised and stored in a password-protected database under the guidelines of the Data Protection Act 1998. Patient records will be kept on paper in the form of the diary card questionnaires and clinical scoring sheets. These will be kept in a locked filing cabinet at Charing Cross Hospital for 10 years in accordance with Imperial College London’s policy.
The chief investigator will preserve the confidentiality of participants taking part in the study and is registered under the Data Protection Act.
Patient details will be anonymised as each participant will be allocated a study number. The allocated study number key code will be kept on a password-protected NHS computer at all sites.
Patient details, including contact information, will be recorded on the paper form. This paper form will be kept in a locked filing cabinet in a locked vascular research office, located in the Section of Vascular Surgery on Floor 4 North at Charing Cross Hospital (university office). The contact details will be discarded once patients have been advised of the findings of the study (within approximately 6–12 months following completion of the study).
All anonymised patient details with the allocated study number used as an identifier will be stored electronically on a password-protected access database on an Imperial College London university computer.
Data and study findings will be presented locally within the hospital, as well as at national and international peer-reviewed presentations.
All participants will be sent reminders by post or contacted over the telephone to attend the follow-up visits. In case if they fail to attend a scheduled visit, QoL questionnaires will be sent by post to be completed.
Statistical analysis
Data will be analysed using SPSS version 24 (IBM, Armonk, NY, USA) STATA 15SE (StataCorp, College Station, TX, USA) or a similar statistical software. Data will be analysed on an intention-to-treat (ITT) basis. Visual testing and Shapiro-Wilk testing will be performed to assess the distribution of the data. For continuous data, if the data is normally distributed mean and standard deviation (SD) will be presented, whereas median and interquartile range (IQR) will be presented if it not normally distributed. For categorical data, frequencies and percentages will be presented. T tests may be conducted if the data is normally distributed, whereas the Mann Whitney U test may be more desirable if data is not normally distributed. A repeated measure analysis of variance (ANOVA) will be used to examine changes in scores from baseline during follow-up. All time-to-event outcomes will be assessed using Kaplan-Meier curves and log-rank tests for group comparison. Chi-squared tests will be performed to compare treatment group proportions. Missing data will be handled with multiple imputation methods.
Cost-effectiveness analysis
Cost-effectiveness analysis for both interventions will be assessed using the cost of the equipment and the time required to perform the procedure, the cost of personnel involved, the cost of theatre usage, time to return to work and QoL gain following the procedure.
Data monitoring, safety and quality control
An adverse event (AE) is an untoward medical occurrence in a patient or clinical study subject, which may or may not be caused by the investigational device. All such events, whether expected or not, should be recorded.
A serious adverse event (SAE) is an untoward and unexpected medical occurrence or effect that results in death or is life-threatening; specifically referring to an event in which the subject was at risk of death at the time of the event. It does not refer to an event which hypothetically might have caused death if it were more severe, requires hospitalisation or prolongation of existing inpatients’ hospitalisation, results in persistent or significant disability or incapacity, or results from a congenital anomaly or birth defect.
All AEs should be reported. Depending on the nature of the event the reporting procedures below should be followed. Any questions concerning AE reporting should be directed to the chief investigator in the first instance.
An SAE form should be completed and sent by fax or email to the chief investigator within 24 h. All SAEs should be reported to the Research Ethical Committee where, in the opinion of the chief investigator, the event was ‘related’ (i.e. resulted from the administration of any of the research procedures) and ‘unexpected’ (i.e. an event that is not listed in the protocol as an expected occurrence).
Reports of related and unexpected SAEs should be submitted within 15 days of the chief investigator becoming aware of the event, using the NRES SAE form for non-IMP studies.
Local investigators should report any SAEs as required by their Local Research Ethics Committee, sponsor and/or Research and Development Office.
In the event of any harm to participants in the trial, Imperial College holds public liability (‘negligent harm’) and clinical trial (‘non-negligent harm’) insurance policies which apply to this trial.
The study will be monitored and audited with Joint Research Compliance Office (JRCO) policy.
Ethical approval and study registration
Ethical approval has been sought from a Regional Research Ethics Committee London (REC Ref: 17/LO/1457). The study has been registered at the ClinicalTrials.gov website (https://clinicaltrials.gov/show/NCT03392753). The study will be conducted in accordance with the recommendations for physicians involved in research on human subjects adopted by the 18th World Medical Assembly, Helsinki 1964 and later revisions.
Discussion
Varicose veins are a very common condition associated with detrimental effects on the QoL of patients [2,3,4]. It has been proven that their management leads to improvement in QoL [2,3,4]. Recently, the newer techniques of MOCA and CAE have been introduced and do not require tumescent anaesthesia or the use of thermal energy. This has led to a reduction in patient discomfort, hematoma formation, and risk of a nerve injury when compared to thermal-based procedures.
The results, so far, of these new techniques have shown to be effective and safe [18, 19]; however, there is little data regarding their efficiency and durability.
Therefore, we have designed this multicentre randomised controlled trial to ascertain which one of these techniques causes less pain. To our knowledge, this is the first trial making the direct comparison between MOCA and CAE.
The aims of this current trial are firstly to compare the pain score between MOCA versus CAE and, secondly, to assess the pain level over the ensuing few days, change in the QoL, VCSS, degree of bruising and inflammation, time to return to normal activities/work, re-intervention rates and cost-effectiveness of each method.
The trial will be conducted across two very different healthcare systems in the UK and Singapore, broadening the population base, characteristics and application of findings.
Trial status
At the time of submission, the study has commenced recruitment of participants in November 2017. The three collaborating centres are Charing Cross Hospital (Imperial College London, UK), Singapore General Hospital and Sengkang General Hospital (Singapore).
Abbreviations
- ABPI:
-
Ankle Brachial Pressure Index
- AE:
-
Adverse events
- AVF:
-
American Venous Forum
- AVVQ:
-
Aberdeen Varicose Vein Questionnaire
- CAE:
-
Cyanoacrylate adhesive injection
- CEAP:
-
Clinical Aetiology Anatomy Pathophysiology
- CIVQ-14:
-
Chronic Venous Insufficiency Questionnaire
- CVD:
-
Chronic venous disease
- DUS:
-
Duplex ultrasound
- EQ5D:
-
EuroQol’s 5-domain Utility Index
- EQ-VAS:
-
EuroQol’s Visual Analogue Scale
- EVLA:
-
Endovenous laser ablation
- GSV:
-
Great saphenous vein
- MOCA:
-
Mechanochemical ablation
- NICE:
-
The National Institute of Clinical Excellence
- NTNT:
-
Non-thermal non-tumescent
- QoL:
-
Quality of life
- RFA:
-
Radiofrequency ablation
- SAE:
-
Serious adverse events
- SFJ:
-
Saphenous-femoral junction
- SPJ:
-
Sapheno-popliteal Junction
- SSV:
-
Small saphenous vein
- VAS:
-
Visual Analogue Scale
- VCSS:
-
Venous Clinical Severity Score
References
Evans CJ, Fowkes FG, Ruckley CV, Lee AJ. Prevalence of varicose veins and chronic venous insufficiency in men and women in the general population: Edinburgh vein study. J Epidemiol Community Health. 1999;53(3):149–53. https://doi.org/10.1136/jech.53.3.149.
Smith JJ, Garratt AM, Guest M, Greenhalgh RM, Davies AH. Evaluating and improving health-related quality of life in patients with varicose veins. J Vasc Surg. 1999;30(4):710–9. https://doi.org/10.1016/S0741-5214(99)70110-2.
MacKenzie RK, Paisley A, Allan PL, Lee AJ, Ruckley CV, Bradbury AW. The effect of long saphenous vein stripping on quality of life. J Vasc Surg. 2002;35(6):1197–203. https://doi.org/10.1067/mva.2002.121985.
Biemans AAM, Kockaert M, Akkersdijk GP, van den Bos RR, de Maeseneer MGR, Cuypers P, et al. Comparing endovenous laser ablation, foam sclerotherapy, and conventional surgery for great saphenous varicose veins. Journal of Vascular Surgery. 2013;58(3):727–734.e1. https://doi.org/10.1016/j.jvs.2012.12.074.
Gloviczki P, Gloviczki ML. Guidelines for the management of varicose veins. Phlebology. 2012;27(Suppl 1(Cvi)):2–9. https://doi.org/10.1258/phleb.2012.012S28.
Marsden G, Perry M, Kelley K, Davies AH, Guideline Development Group. Diagnosis and management of varicose veins in the legs: summary of NICE guidance. BMJ. 2013;347:f4279. https://doi.org/10.1136/BMJ.F4279.
National Clinical Guideline Centre (UK). Varicose Veins in the Legs: The Diagnosis and Management of Varicose Veins. London: National Institute for Health and Care Excellence (UK); 2013. (NICE Clinical Guidelines, No. 168.). (n.d.). Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK264166/. https://www.nice.org.uk/guidance/cg168/resources/varicose-veins-diagnosis-and-management-pdf-35109698485957.
Subramonia S, Lees T. Randomized clinical trial of radiofrequency ablation or conventional high ligation and stripping for great saphenous varicose veins. Br J Surg. 2010;97(3):328–36. https://doi.org/10.1002/bjs.6867.
Rasmussen LH, Lawaetz M, Bjoern L, Vennits B, Blemings A, Eklof B. Randomized clinical trial comparing endovenous laser ablation, radiofrequency ablation, foam sclerotherapy and surgical stripping for great saphenous varicose veins. Br J Surg. 2011;98(8):1079–87. https://doi.org/10.1002/bjs.7555.
Tolva VS, Cireni LV, Bianchi PG, Almarosa L, Keller GC, Casana RM. Radiofrequency ablation of the great saphenous vein with the ClosureFAST TM procedure: mid-term experience on 400 patients from a single centre. n.d.; https://doi.org/10.1007/s00595-012-0296-4.
Goode S, Chowdhury A, Crockett M, Beech A, Simpson R, Richards T, Braithwaite B. Laser and radiofrequency ablation study (LARA study): a randomised study comparing radiofrequency ablation and Endovenous laser ablation (810nm). Eur J Vasc Endovasc Surg. 2010;40(2):246–53. https://doi.org/10.1016/j.ejvs.2010.02.026.
Anwar MA, Lane TRA, Davies AH, Franklin IJ. Complications of radiofrequency ablation of varicose veins. Phlebology. 2012;27(Suppl 1):34–9. https://doi.org/10.1258/phleb.2012.012S21.
Merchant RF, Pichot O. Long-term outcomes of endovenous radiofrequency obliteration of saphenous reflux as a treatment for superficial venous insufficiency. J Vasc Surg. 2005;42(3):1–9. https://doi.org/10.1016/j.jvs.2005.05.007.
Rasmussen L, Lawaetz M, Bjoern L, Blemings A, Eklof B. Randomized clinical trial comparing endovenous laser ablation and stripping of the great saphenous vein with clinical and Duplex outcome after 5 years. J Vasc Surg. 2013;58(2):421–6. https://doi.org/10.1016/j.jvs.2012.12.048.
Dexter D, Kabnick L, Berland T, Jacobowitz G, Lamparello P, Maldonado T, et al. Complications of endovenous lasers. Phlebology. 2012; https://doi.org/10.1258/phleb.2012.012S18.
Elias SR, Raines JK. Mechanochemical tumescentless endovenous ablation: final results of the initial clinical trial. Phlebology. 2012;27(3):67–72. https://doi.org/10.1258/phleb.2011.010100.
Almeida JI, Javier JJ, Mackay E, Bautista C, Proebstle TM. First human use of cyanoacrylate adhesive for treatment of saphenous vein incompetence. J Vasc Surg Venous Lymphat Disord. 2013;1(2):174–80. https://doi.org/10.1016/j.jvsv.2012.09.010.
Morrison N, Gibson K, McEnroe S, Goldman M, King T, Weiss R, et al. Randomized trial comparing cyanoacrylate embolization and radiofrequency ablation for incompetent great saphenous veins (VeClose). J Vasc Surg. 2015;61(4):985–94. https://doi.org/10.1016/j.jvs.2014.11.071.
Lane T, Bootun R, Dharmarajah B, Lim CS, Najem M, Renton S, et al. A multi-centre randomised controlled trial comparing radiofrequency and mechanical occlusion chemically assisted ablation of varicose veins—final results of the Venefit versus Clarivein for varicose veins trial. Phlebology. 2017;32(2):89–98. https://doi.org/10.1177/0268355516651026.
Mechanochemical ablation for varicose veins; Interventional procedures guidance. 2016. Retrieved from https://www.nice.org.uk/guidance/ipg557/resources/endovenous-mechanochemical-ablation-for-varicose-veins-pdf-1899871992297925.
Cyanoacrylate glue occlusion for varicose veins; Interventional procedures guidance. 2015. Retrieved from https://www.nice.org.uk/guidance/ipg526/resources/cyanoacrylate-glue-occlusion-for-varicose-veins-pdf-1899871809219781
Van Eekeren RRJP, Boersma D, Konijn V, De Vries JPPM, Reijnen MMJP. Postoperative pain and early quality of life after radiofrequency ablation and mechanochemical endovenous ablation of incompetent great saphenous veins. J Vasc Surg. 2013;57(2):445–50. https://doi.org/10.1016/j.jvs.2012.07.049.
Almeida JI, Javier JJ, Mackay E, Bautista C, Proebstle TM. First human use of cyanoacrylate adhesive for treatment of saphenous vein incompetence. J Vasc Surg. 2013;1:174–80. https://doi.org/10.1016/j.jvsv.2012.09.010.
Shepherd AC, Gohel MS, Brown LC, Metcalfe MJ, Hamish M, Davies AH. Randomized clinical trial of VNUS ClosureFAST radiofrequency ablation versus laser for varicose veins. Br J Surg. 2010;97:810–8.
Huskisson E. Measurement of pain. Lancet. 1999;79(2):231–51. https://doi.org/10.1016/S0140-6736(74)90884-8.
Almeida J, Mackay E, Javier J, Mauriello J, Raines J. Saphenous laser ablation at 1470 nm targets the vein wall, not blood. Vasc Endovasc Surg. 2009;43(5):467–72. https://doi.org/10.1177/1538574409335916.
Acknowledgements
Infrastructure support for this work was provided by the NIHR Imperial Biomedical Research Centre.
Availability of data and materials
Anonymised study data will be available on request and will be uploaded to the Imperial College London Secure Research Server. All data analyses and manuscripts produced will be available on request.
Confidentiality
Indemnity
Imperial College London holds negligent harm and non-negligent harm insurance policies which apply to this study.
Sponsor
Imperial College London will act as the main sponsor for this study. Delegated responsibilities will be assigned to the NHS trusts taking part in this study.
Consent to participate
Consent to enter the study will be sought from each participant only after a full explanation has been given and an information leaflet offered and time allowed for consideration. The co-investigators at each recruiting canter will obtain written consent from all participants. The right of the participant to refuse to participate without giving reasons will be respected. After the participant has entered the study, the clinician remains free to give alternative treatment to that specified in the protocol at any stage if they feel that it is in the participant’s best interest, but the reasons for doing so will be recorded. In these cases, the participants remain within the study for the purposes of follow-up and data analysis. All participants are free to withdraw at any time from the protocol treatment without giving reasons and without prejudicing further treatment.
Author information
Authors and Affiliations
Contributions
All authors drafted the study protocol and design. RB, TL and AD participated in the design of the study. AB coordinates the project and drafted the manuscript. TL and AB perform the statistical analysis. AD supervises the project. AB recruits the patients and drafted the manuscript. TYT will recruit patients from the two hospitals in Singapore. All authors will read and approve the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
National Research Ethics Service approval: Regional Committee London: (REC Ref: 17/LO/1457).
Consent for publication
Consent for publication will be sought from each participant at the time of taking consent for participation.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) 2013 Checklist: recommended items to address in a clinical trial protocol and related documents. (DOC 123 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Belramman, A., Bootun, R., Tang, T.Y. et al. Mechanochemical ablation versus cyanoacrylate adhesive for the treatment of varicose veins: study protocol for a randomised controlled trial. Trials 19, 428 (2018). https://doi.org/10.1186/s13063-018-2807-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-018-2807-0