
Abstract
Background
Acute respiratory failure (ARF) is the leading reason for intensive care unit (ICU) admission in immunocompromised patients. High-flow nasal oxygen (HFNO) therapy is an alternative to standard oxygen. By providing warmed and humidified gas, HFNO allows the delivery of higher flow rates via nasal cannula devices, with FiO2 values of nearly 100%. Benefits include alleviation of dyspnea and discomfort, decreased respiratory distress and decreased mortality in unselected patients with acute hypoxemic respiratory failure. However, in preliminary reports, HFNO benefits are controversial in immunocompromised patients in whom it has never been properly evaluated.
Methods/design
This is a multicenter, open-label, randomized controlled superiority trial in 30 intensive care units, part of the Groupe de Recherche Respiratoire en Réanimation Onco-Hématologique (GRRR-OH). Inclusion criteria will be: (1) adults, (2) known immunosuppression, (3) ARF, (4) oxygen therapy ≥ 6 L/min, (5) written informed consent from patient or proxy. Exclusion criteria will be: (1) imminent death (moribund patient), (2) no informed consent, (3) hypercapnia (PaCO2 ≥ 50 mmHg), (4) isolated cardiogenic pulmonary edema, (5) pregnancy or breastfeeding, (6) anatomical factors precluding insertion of a nasal cannula, (7) no coverage by the French statutory healthcare insurance system, and (8) post-surgical setting from day 1 to day 6 (patients with ARF occurring after day 6 of surgery can be included).
The primary outcome measure is day-28 mortality. Secondary outcomes are intubation rate, comfort, dyspnea, respiratory rate, oxygenation, ICU length of stay, and ICU-acquired infections.
Based on an expected 30% mortality rate in the standard oxygen group, and 20% in the HFNO group, error rate set at 5%, and a statistical power at 90%, 389 patients are required in each treatment group (778 patients overall). Recruitment period is estimated at 30 months, with 28 days of additional follow-up for the last included patient.
Discussion
The HIGH study will be the largest multicenter, randomized controlled trial seeking to demonstrate that survival benefits from HFNO reported in unselected patients also apply to a large immunocompromised population.
Trial registration
ClinicalTrials.gov, ID: NCT02739451. Registered on 15 April 2016.
Similar content being viewed by others

Background
Acute respiratory failure (ARF) is the leading reason for intensive care unit (ICU) admission of immunocompromised patients [1,2,3,4,5,6]. Mortality has decreased dramatically in this population in recent years, for several reasons. Management strategies for the underlying conditions have benefited from a number of innovations such as steroid-sparing agents, watch-and-wait approaches, and targeted therapies [7, 8]. Early ICU admission to permit the use of non-invasive diagnostic and therapeutic strategies has increased survival [1, 9,10,11]. Finally, the introduction of other oxygenation strategies has improved the management of respiratory dysfunction (Table 1).
Oxygen therapy is the first-line treatment in hypoxemic patients. Oxygen can be delivered using low-flow devices (up to 15 L/min) such as nasal cannulas, non-rebreathing masks, and bag-valve masks. The fraction of inspired oxygen (FiO2) obtained using these devices varies with the patient’s breathing pattern, peak inspiratory flow rate, delivery system, and mask characteristics. Maximum flow rates are limited in part by the inability of these devices to heat and humidify gas at high flows. Also, if the patient has a high inspiratory flow rate, the amount of entrained room air is large and dilutes the oxygen, thereby lowering the FiO2.
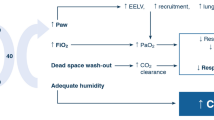
Over the past two decades, devices that deliver heated and humidified oxygen at high flow rates through a nasal cannula were developed as an alternative to low/medium-flow devices. High-flow nasal oxygen (HFNO) delivers oxygen flow rates of up to 60 L/min. An air/oxygen blender is connected via an active heated humidifier to a nasal cannula and allows FiO2 adjustment independently from the flow rate. Compared to other devices, HFNO provides a number of physiological benefits including greater comfort and tolerance, more effective oxygenation under some circumstances and breathing pattern improvements with an increase in tidal volume and decreases in respiratory rate and dyspnea (Tables 2 and 3). These benefits are broadening the indications of HFNO, which has now been evaluated and used to treat hypoxemic respiratory failure, to improve oxygenation for pre-intubation, and to treat patients after surgery or after extubation (Table 4). Moreover, recent high-quality randomized controlled trials (RCTs) have confirmed previous preliminary results [12,13,14]. Nevertheless, controlled studies in specific patient populations, such as immunocompromised patients, are needed to confirm that HFNO is clinically superior over other methods, to evaluate effects on survival, and to determine the optimal indications of HFNO compared to other modalities such as standard oxygen therapy and non-invasive ventilation (NIV).
Among patients with ARF, those with immunosuppression have higher mortality rates compared to unselected patients. The use of endotracheal mechanical ventilation is associated with higher mortality in immunocompromised patients. Therefore, management techniques that decrease the need for intubation may hold promise for decreasing mortality [53,54,55,56].
Four studies evaluated the feasibility and safety of HFNO in immunocompromised patients with ARF. In a retrospective, single-center study reported in 2013, the feasibility of HFNO was evaluated in 45 patients with hematological malignancies [57]. Of the 45 patients, 15 recovered without intubation (33%); their hospital mortality rate was 2/15 (13%), compared to 26/30 (87%) of the patients who failed HFNO and required intubation. HFNO failure was significantly associated with bacterial pneumonia as the cause of ARF. In a single-centre study of patients with solid tumors reported in 2011, of 183 patients taken at random from the institutional database, 132 (72%) had received HFNO in the ICU to treat hypoxia [58]. Among them, 41% improved and 44% remained stable while on HFNO, whereas 15% declined. A 2013 report describes a study in 30 patients with advanced cancer and persistent dyspnea that used a randomized design to compare the physiological effects of HFNO vs. bi-level positive airway pressure (BiPAP) for 2 h [59]. Both treatments similarly improved the dyspnea, as assessed using a Visual Analog Scale (VAS) and the modified Borg scale, and non-significantly diminished the respiratory rate. Oxygen saturation improved only with HFNO. Neither technique induced major adverse effects. The last study, published in 2015, evaluated HFNO for treating ARF requiring ICU admission in 37 lung transplant recipients [60]. HFNO proved feasible and safe and decreased the absolute risk of intubation by 29%, with a number-needed-to-treat to avoid one intubation of three. Last, in a study of 50 Do-Not-Intubate patients with hypoxemic respiratory distress, including a third of immunocompromised patients, HFNO allowed an improvement in oxygenation and decreased respiratory rate [61].
Four studies assessed HFNO efficacy in immunocompromised patients with ARF. The first study, by Mokart et al., analyzed a retrospective cohort of 178 patients with cancer and ARF (O2 > 9 L/min), including 76 (43%) treated with NIV + HFNO, 74 (42%) with NIV + low/medium-flow O2, 20 (11%) with HFNO alone, and 8 with low/medium-flow O2 alone [62]. NIV + HFNO was associated with lower mortality (37% vs. 52% in remaining patients, p = 0.04) and was independently associated with lower day-28 survival in a propensity-score analysis. Second, in a substudy of data from our recent iVNIctus RCT of early NIV in immunocompromised patients with ARF [63,64,65], 141/374 (38%) patients received HFNO, and either NIV or low/medium-flow oxygen was used in the other patients. To allow accurate adjustment, we built a propensity score using variables available at ICU admission. Intubation rate and day-28 mortality were not significantly different in the HFNO arm compared to the NIV or low/medium-flow oxygen arm. Third, in 115 immunocompromised patients with ARF, 60 (52%) were treated with HFN0 alone and 55 (48%) with NIV as first-line therapy with 30 patients (55%) receiving HFNO and 25 patients (45%) standard oxygen between NIV sessions [66]. The rates of intubation and 28-day mortality were higher in patients treated with NIV than with HFNO (55 vs. 35%, p = 0.04, and 40 vs. 20%, p = 0.02, respectively). Using propensity score-matched analysis, NIV was associated with mortality. Using multivariate analysis, NIV was independently associated with intubation and mortality. Last, in a post-hoc analysis of the FLORALI study that only included immunocompromised patients, 8 (31%) of 26 HFNO patients, 13 (43%) of 30 patients treated with standard oxygen, and 17 (65%) of 26 patients treated with NIV required intubation at 28 days (p = 0·04). Odds ratios for intubation did not differ, however, between HFNO patients and those receiving standard oxygen only [67]. Last, in the Efraim study that included 1611 immunocompromized patients with acute respiratory failure, the use of HFNO had an effect on intubation rate but not on mortality, whereas, failure to identify ARF etiology was associated with increased intubation rate and mortality [68].
Although the effects of HFNO have varied across studies, the data establish that this treatment modality is feasible and safe in immunocompromised patients. They also demonstrate that outcomes with HFNO are at least as good as with other oxygen therapy methods in this population. Thus, they warrant further trials to determine whether HFNO improves survival in unselected immunocompromised patients with hypoxemic ARF. Immunocompromised patients have specific treatment needs, as shown by their two-fold higher mortality rate after intubation compared to other patients. Data on HFNO in immunocompromised patients are conflicting.
We therefore designed the present RCT (HIGH). This RCT is a superiority study of HFNO vs. other oxygenation strategies (low/medium-flow oxygen) in immunocompromised patients requiring oxygen. The primary endpoint is day-28 survival. The patients will be recruited at 31 centers belonging to the Groupe de Recherche Respiratoire en Réanimation Onco-Hématologique (GRRR-OH), a research network that specializes in the management of critically ill immunocompromised patients and has a particularly high level of expertise in respiratory care strategies. The control group will receive low/medium-flow oxygen as deemed appropriate by the physician since the recent large iVNIctus trial by our group did not show any superiority of NIV on intubation rates or survival. The experimental group will receive continuous HFNO at any time after ICU admission, for pre-oxygenation before intubation, after extubation, and for any ICU procedure that might induce hypoxemia). HFNO will not be used in the control group.
Methods/design
Design and settings
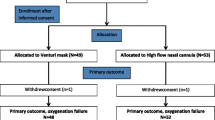
The HIGH trial is a prospective, multicenter, open-label RCT comparing HFNO vs. other oxygenation strategies (low/medium-flow oxygen) in immunocompromised patients requiring oxygen. The study hypothesis is that early HFNO decreases mortality on day 28 after randomization in immunocompromised patients requiring ICU admission for ARF. Figure 1 shows an adapted version of the SPIRIT Figure for the trial (Additional file 1).

SPIRIT checklist
Ethical aspects
The study was approved by the local Independent Ethic Committee (Comité de Protection des Personnes CPP Ile de France IV, Saint Louis on 28 March 2016, number 2016/08), the French health authorities (AFSSAPS) on 14 March 2016, number EudraCT 2016-A00220-51. The University Hospital of Paris (AP-HP) and, by delegation, the Clinical Research and Development Department (DRCD) is the sponsor of the trial (sponsor code: P150912/IDRCB No: 2016-A00220-51). Informed consent will be obtained from each participant.
Participating intensive care units
All participating centers belong to the GRRR-OH. All these centers have previously taken part in observational studies, surveys, or therapeutic trials. They all have high case-volumes of patients with immune deficiencies due to immunosuppressive drugs, solid-organ transplantation, malignancies, or systemic diseases. Although they are specialized in oncology and hematology, they also admit high volumes of patients with systemic diseases, solid-organ transplants and other immunosuppression.
Study population
Eligible patients are immunocompromised patients who are admitted to the ICU and need oxygen supplementation (of at least 6 L/min) at any stage of their ICU stay. All randomized patients will be included in the full set of analysis (intent-to-treat basis).
To be randomized patients should fulfill all the following inclusion criteria (1) adult (age ≥ 18 years), (2) known immunosuppression defined as one or more of the following: immunosuppressive drugs/long-term (> 3 months) or high-dose (> 0.5 mg/kg/day) steroids, solid-organ transplant, solid tumor having required cancer care in the last 5 years, hematological malignancy or primary immune deficiency, (3) ICU admission for ARF, (4) need for oxygen therapy ≥ 6 L/min, and (5) written informed consent from the patient or proxy (if present) before inclusion or once possible when patient has been included in a context of emergency.
Exclusion criteria were: (1) imminent death (moribund patients), (2) refusal of study participation or to pursue the study by the patient, (3) hypercapnia with a formal indication for NIV (PaCO2 ≥ 50 mmHg, formal indication for NIV), (4) isolated cardiogenic pulmonary edema (formal indication for NIV). Patients with pulmonary edema associated with another ARF etiology can be included, (5) pregnancy or breastfeeding, (6) anatomical factors precluding the use of a nasal cannula, (7) absence of coverage by the French statutory healthcare insurance system, and (8) post-surgical setting from day 1 to day 6 (patients with ARF occurring after day 6 of surgery can be included).
Randomization
Randomization will be stratified on three factors, measured at study inclusion, namely: (1) time since ICU admission, segregating day 0 (calendar date of ICU admission), day 1, day 2 vs. ≥ day 3; (2) hypoxemia severity, segregating oxygen flow < vs. ≥ 9 L to reach SpO2 ≥ 95% at randomization, and (3) shock, based on the administration of catecholamine. Thus, analysis could consider treatment-by-subset interaction on such strata.
Randomization will be achieved using an electronic system incorporated in the eCRF and R software (http://www.R-project.org/). The impact of the intervention will be assessed at the patient level. The randomization unit is the center. Randomization will be centralized on a web site to ensure allocation concealment at the trial statistical center. Patients will be randomized into two parallel groups in a 1:1 ratio. Randomization will be stratified (see above), resulting in eight different randomization lists that will be pre-specified and balanced through the use of permutation blocks of fixed size that will not be disclosed to the local investigators, to ensure allocation concealment and to avoid all risk of bias in patient selection.
Study interventions
This open RCT will compare two oxygenation strategies.
Standard oxygen as the usual care (control group)
Patients in the control group will receive the best standard of care, according to the usual practice of the local intensivists and primary-care physicians. Oxygen therapy will be delivered using any device or combination of devices that are part of usual care: nasal oxygen, and mask with or without a reservoir bag and with or without the Venturi system. Oxygen settings are set to target a SpO2 ≥ 95%. HFNO will not be used in the control group. NIV will not be used at all in this trial, unless patients develop hypercapnia or pulmonary edema throughout the ICU stay, for the time they meet these conditions. ICU discharge will be allowed when patients will meet the ability to maintain SpO2 ≥ 95% with less than 6 L/min oxygen.
High-flow nasal oxygen (intervention group)
Patients in the HFNO group will receive the best standard of care, according to the usual practice of the local intensivists and primary physicians, with one exception: supplemental oxygen will be provided only by continuous HFNO. HFNO will be initiated at a flow rate of 50 L/min and 100% FiO2. If the target SpO2 is not reached, the flow rate will be increased to 60 L/min. Then, FiO2 will be tapered to target a SpO2 ≥ 95%. The minimal flow rate within the first 3 days will be 50 L/min. In patients who require intubation, HFNO will be used during laryngoscopy and immediately after extubation. Also, HFNO will be used before, during, and after all ICU procedures. Patients with discomfort due to HFNO will have their flow rate decreased until the discomfort resolves. If the nasal prongs generate significant discomfort or skin breakdown, a Venturi mask will be used instead until HFNO can be used again; except in this situation, standard oxygen will be used in the intervention group. NIV will, however, be used in the same conditions than in the control group.
HFNO will be stopped based on clinical criteria (improvement of clinical signs of respiratory distress), PaO2/FiO2 > 300, and ability to maintain SpO2 ≥ 95% with less than 6 L/min of standard oxygen (allowing ICU discharge as HFNO may not be available in the wards).
Data collection and follow-up
Evaluation at study inclusion (T0)
The evaluation at study inclusion will include patient’s characteristics, underlying disease, associated organ dysfunction, investigations usually performed at ICU admission in immunocompromised patients with ARF, and ARF etiology.
Evaluations throughout study participation
Evaluations performed throughout study participation will include physiological variables including respiratory and ventilation parameters (respiratory rate, SpO2, oxygen flow and/or FiO2), blood gases and chest x-ray (the worst values will be recorded). Results of investigations, ICU-acquired infections and data on oxygenation tolerance and efficacy as well as on comfort will be also collected.
ICU-acquired infections are defined as any new-onset infection starting more than 48 h after ICU admission for which the clinical team started a new antibiotic regimen. Every single infection diagnosis will be made using Centers for Disease Control and Prevention definitions [69].
Evaluation at the end of study participation
Evaluations performed at the end of study participation will consist of mortality on day 28, need for intubation, ICU and hospital lengths of stay and ICU-acquired infections. All elements allowing to record secondary endpoints will be collected.
Organization of the trial
Funding and support
The HIGH trial is promoted by the Assistance Publique – Hôpitaux de Paris and supported by a grant from the French Ministry of Health (Programme Hospitalier de Recherche Clinique 2012; AOM12456).
Coordination and implementation of the trial
Each medical and paramedical team in the 31 participating ICUs were trained in the protocol and data collection using an electronic case-record form during formal meetings prior to screening and inclusion. The electronic case-record form was developed with CleanWEBTM, a centralized, secure, interactive, web-response system accessible from each study center, provided and managed by Telemedicine Technologies.
Local physicians and clinical research assistants in each participating ICU are responsible for daily screening and inclusion of patients, compliance with protocol, availability of data requested for the trial and completion of the electronic case-record form. In accordance with French law, the electronic case-record form and database were validated by the appropriate committees (Comité Consultatif sur le Traitement de l’Information en matière de Recherche dans le domaine de la Santé; Commission Nationale de l’Informatique et des Libertés).
Interim analysis
One interim analysis by an independent Data Safety and Monitoring Board is planned after the occurrence of 100 deaths. The Data Safety and Monitoring Board will be blinded to allocation of groups and may decide premature termination of the study. The board consists of one methodologist, one pulmonologist, and one intensivist. Data are blindly analyzed but unblinding is possible on request of the Data Safety and Monitoring Board. An extraordinary meeting may be requested by the principal investigator or the methodologist, in the case of unexpected events that might affect continuation of the protocol.
Blinding
Given the nature of the interventions, physicians, nurses, and patients cannot be blinded for the randomized interventions. The analysis will be blinded to allocation of groups.
Study outcomes
Primary endpoint
The primary endpoint of this trial is day-28 mortality.
Secondary endpoints
The secondary endpoints are: intubation rate (proportion of patients requiring invasive mechanical ventilation) on day 28, patient comfort VAS, dyspnea (VAS and Borg scale), respiratory rate, oxygenation (based on the lowest SpO2 value and on PaO2/FiO2 from day 1 to day 3, ICU stay length, incidence of ICU-acquired infections.
Statistical methods
All statistical analyses will be performed using SAS (SAS Inc., Cary, NC, USA) and R (http://www.R-project.org/) software.
Sample size calculation
Based on a 30% day-28 mortality rate in usual-care oxygen group, and a 20% day-28 mortality rate in the HFNO group, with α set at 5%, to obtain a 90% power for demonstrating superiority for the primary outcome, we need 778 patients (389 in each group).
Recruitment is expected to take 30 months, and 28 additional days will be required for follow-up.
Interim analyses
One interim analysis will be performed once 100 deaths will have been observed. Due to inflation of type I error consideration, it will use the Haybittle-Peto boundary, that is a p value threshold of 0.001 for the interim analysis (while the terminal analysis will use a threshold of 0.05, as scheduled in the sample size computation). Moreover, to get insight in the difference across arms in terms of futility or efficacy, the Bayesian posterior probability of the 28-day mortality rate and of the log odds ratio will be computed, using a uniform non-informative prior. The final analysis will be started after inclusion of the planned number of patients.
Methodology of the statistical analysis
The main comparison based on the intention-to-treat principle will compare the intervention arm to the control arm on the full-set of randomized patients. The primary hypothesis is superiority of the NIV in terms of 28-day mortality (primary outcome). For all secondary outcomes, our hypothesis is that HFNO is superior over standard oxygen, with two-sided p values for comparison tests. Secondary and exploratory comparisons of the primary endpoint will look for treatment-by-covariate interactions according to the subsets defined above. Finally, a per-protocol analysis will be performed.
Missing values and outliers
Missing values for the main outcome measure are not expected to be observed; nevertheless, in case of occurrence, they will be handled using time-to-event methods in which each patient contributes to the estimate of failure time distribution until they are lost-to-follow up or withdrawn from the study using competing-risks estimates. Missing values for predictors will be imputed using multiple imputation techniques.
Analysis of the primary outcome
The main endpoint is binary, as all patients will be followed until day 28, at which time they will be classified as alive or dead. The relative risk of hospital death in the experimental vs. the control arm will be estimated to assess the effectiveness of the intervention, with 95% confidence interval. Analyses adjusted on potential confounders will be performed. Intervention-by-subsets interactions will be tested using Gail and Simon statistics. In case of significant interaction, subset analyses will be performed on each subset.
Analysis of the secondary outcomes
Competing-risk endpoints (ICU-acquired events including intubation, ICU-acquired infection) will be analyzed using competing-risk methods. Specifically, cumulative incidences of the event of interest will be estimated, taking into account the competition between death or discharge alive from the ICU and the event of interest, then compared using the Gray test. Adjustment for potential confounders will be based on cause-specific Cox models. ICU length of stay will be analyzed overall and in survivors and dead patients, separately. The former analysis will be based on Kaplan-Meier estimates while the later on the competing-risk estimator, as described above. Analyses of longitudinal outcomes (oxygenation, dyspnea, patient’s comfort) will be based on joint models, taking into account the right censoring of the data.
Discussion
ARF remains the most frequent and challenging life-threatening event in patients with hematological malignancies. In patients with prolonged neutropenia (acute leukemia or bone marrow transplant recipients), respiratory events occur in up to half of cases, of which a further half are complicated by ARF. Despite a recent improvement in survival, intubation and subsequent invasive mechanical ventilation remains associated with high mortality in immunocompromised patients with ARF. In recent studies, mortality after intubation was 60% in hematological patients and 40% in immunocompromised patients. In that setting, any strategy that could prevent intubation and subsequent increase in mortality could be of benefit.
HFNO has been associated with an increase survival for immunocompetent patients managed in the ICU for a hypoxemic ARF, and with a decrease in intubation rate in the most hypoxemic patients. Nevertheless, data are scarce in specific patient populations, such as immunocompromised patients, who are at high risk of intubation when presenting with ARF. Clearly, data are needed to confirm that HFNO is clinically superior over other methods in immunocompromised patients. This fully justifies the HIGH trial.
As a consequence of the negative result of our recent iVNIctus multicentre RCT that did not show a benefit of NIV on mortality nor on intubation in immunocompromised patients with ARF, we have decided that NIV would not delivered in a systematic way to the patients included in the HIGH trial. In addition, recent data from an ancillary study of the FLORALI trial suggests that intubation rate and mortality were higher in patients treated with NIV than in those treated with HFNO. However, clinicians in charge will be allowed to deliver NIV to patients with a well-established indication of NIV, such as cardiogenic pulmonary edema and hypercapnic ARF.
We expect the HIGH trial to assess an oxygenation management strategy including HFNO. We hypothesize that mortality will be lower in patient receiving HFNO, possibly in association with a reduction of the intubation rate. We also expect the HIGH trial to analyze the factors that predict intubation in immunocompromised patients with ARF.
Trial status
Enrollment is ongoing, having started on May 2016. The first interim analysis was conducted on 13 March 2017, and the Data Safety and Monitoring Board recommended that the study be continued. On 13 November 2017, 686 patients were included in the trial. Enrollment is expected to be completed in February 2018.
Abbreviations
- ARF:
-
Acute respiratory failure
- GRRR-OH:
-
Groupe de Recherche Respiratoire en Réanimation Oncohématologique
- HFNO:
-
High-flow nasal oxygen
- ICU:
-
Intensive care unit
- NIV:
-
Non-invasive ventilation
References
Dumas G, Geri G, Montlahuc C, et al. Outcomes in critically ill patients with systemic rheumatic disease: a multicenter study. Chest. 2015;2015(21):14–3098.
Faguer S, Ciroldi M, Mariotte E, et al. Prognostic contributions of the underlying inflammatory disease and acute organ dysfunction in critically ill patients with systemic rheumatic diseases. Eur J Intern Med. 2013;24(3):e40–4.
Soares M, Toffart AC, Timsit JF, et al. Intensive care in patients with lung cancer: a multinational study. Ann Oncol. 2014;25(9):1829–35.
Azoulay E, Lemiale V, Mokart D, et al. Acute respiratory distress syndrome in patients with malignancies. Intensive Care Med. 2014;40(8):1106–14.
Azoulay E, Pene F, Darmon M, et al. Managing critically Ill hematology patients: time to think differently. Blood Rev. 2015;2015(26):00030–2.
Canet E, Osman D, Lambert J, et al. Acute respiratory failure in kidney transplant recipients: a multicenter study. Crit Care. 2011;15(2):R91.
Murphy G, Lisnevskaia L, Isenberg D. Systemic lupus erythematosus and other autoimmune rheumatic diseases: challenges to treatment. Lancet. 2013;382(9894):809–18.
Guillevin L, Pagnoux C, Karras A, et al. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med. 2014;371(19):1771–80.
Hilbert G, Gruson D, Vargas F, et al. Noninvasive ventilation in immunosuppressed patients with pulmonary infiltrates, fever, and acute respiratory failure. N Engl J Med. 2001;344(7):481–7.
Azoulay E, Mokart D, Lambert J, et al. Diagnostic strategy for hematology and oncology patients with acute respiratory failure: randomized controlled trial. Am J Respir Crit Care Med. 2010;182(8):1038–46.
Mokart D, Lambert J, Schnell D, et al. Delayed intensive care unit admission is associated with increased mortality in patients with cancer with acute respiratory failure. Leuk Lymphoma. 2013;54(8):1724–9.
Dewan NA, Bell CW. Effect of low flow and high flow oxygen delivery on exercise tolerance and sensation of dyspnea. A study comparing the transtracheal catheter and nasal prongs. Chest. 1994;105(4):1061–5.
Frat JP, Thille AW, Mercat A, et al. High-flow oxygen through nasal cannula in acute hypoxemic respiratory failure. N Engl J Med. 2015;372(23):2185–96.
Stephan F, Barrucand B, Petit P, et al. High-flow nasal oxygen vs noninvasive positive airway pressure in hypoxemic patients after cardiothoracic surgery: a randomized clinical trial. JAMA. 2015;313(23):2331–9.
Campbell EJ, Baker MD, Crites-Silver P. Subjective effects of humidification of oxygen for delivery by nasal cannula. A prospective study. Chest. 1988;93(2):289–93.
Chanques G, Constantin JM, Sauter M, et al. Discomfort associated with underhumidified high-flow oxygen therapy in critically ill patients. Intensive Care Med. 2009;35(6):996–1003.
Wettstein RB, Shelledy DC, Peters JI. Delivered oxygen concentrations using low-flow and high-flow nasal cannulas. Respir Care. 2005;50(5):604–9.
Wagstaff TA, Soni N. Performance of six types of oxygen delivery devices at varying respiratory rates. Anaesthesia. 2007;62(5):492–503.
Vargas F, Saint-Leger M, Boyer A, Bui NH, Hilbert G. Physiologic effects of high-flow nasal cannula oxygen in critical care subjects. Respir Care. 2015;2015(5):03814.
Itagaki T, Okuda N, Tsunano Y, et al. Effect of high-flow nasal cannula on thoraco-abdominal synchrony in adult critically ill patients. Respir Care. 2014;59(1):70–4.
Corley A, Caruana LR, Barnett AG, Tronstad O, Fraser JF. Oxygen delivery through high-flow nasal cannulae increase end-expiratory lung volume and reduce respiratory rate in post-cardiac surgical patients. Br J Anaesth. 2011;107(6):998–1004.
Sztrymf B, Messika J, Bertrand F, et al. Beneficial effects of humidified high flow nasal oxygen in critical care patients: a prospective pilot study. Intensive Care Med. 2011;37(11):1780–6.
Sztrymf B, Messika J, Mayot T, Lenglet H, Dreyfuss D, Ricard JD. Impact of high-flow nasal cannula oxygen therapy on intensive care unit patients with acute respiratory failure: a prospective observational study. J Crit Care. 2012;27(3):324. e9-13
Dysart K, Miller TL, Wolfson MR, Shaffer TH. Research in high flow therapy: mechanisms of action. Respir Med. 2009;103(10):1400–5.
Parke RL, Eccleston ML, McGuinness SP. The effects of flow on airway pressure during nasal high-flow oxygen therapy. Respir Care. 2011;56(8):1151–5.
Berk JL, Lenner KA, McFadden ER Jr. Cold-induced bronchoconstriction: role of cutaneous reflexes vs. direct airway effects. J Appl Physiol (1985). 1987;63(2):659–64.
Fontanari P, Burnet H, Zattara-Hartmann MC, Jammes Y. Changes in airway resistance induced by nasal inhalation of cold dry, dry, or moist air in normal individuals. J Appl Physiol (1985). 1996;81(4):1739–43.
Chanques G, Riboulet F, Molinari N, et al. Comparison of three high flow oxygen therapy delivery devices: a clinical physiological cross-over study. Minerva Anestesiol. 2013;79(12):1344–55.
Greenspan JS, Wolfson MR, Shaffer TH. Airway responsiveness to low inspired gas temperature in preterm neonates. J Pediatr. 1991;118(3):443–5.
Chikata Y, Izawa M, Okuda N, et al. Humidification performance of two high-flow nasal cannula devices: a bench study. Respir Care. 2014;59(8):1186–90.
Salah B, Dinh Xuan AT, Fouilladieu JL, Lockhart A, Regnard J. Nasal mucociliary transport in healthy subjects is slower when breathing dry air. Eur Respir J. 1988;1(9):852–5.
Negus VE. Humidification of the air passages. Thorax. 1952;7(2):148–51.
Groves DS, Durbin CG Jr. Tracheostomy in the critically ill: indications, timing and techniques. Curr Opin Crit Care. 2007;13(1):90–7.
Parke R, McGuinness S, Eccleston M. Nasal high-flow therapy delivers low level positive airway pressure. Br J Anaesth. 2009;103(6):886–90.
Locke RG, Wolfson MR, Shaffer TH, Rubenstein SD, Greenspan JS. Inadvertent administration of positive end-distending pressure during nasal cannula flow. Pediatrics. 1993;91(1):135–8.
Ritchie JE, Williams AB, Gerard C, Hockey H. Evaluation of a humidified nasal high-flow oxygen system, using oxygraphy, capnography and measurement of upper airway pressures. Anaesth Intensive Care. 2011;39(6):1103–10.
Volsko TA, Fedor K, Amadei J, Chatburn RL. High flow through a nasal cannula and CPAP effect in a simulated infant model. Respir Care. 2011;56(12):1893–900.
Riera J, Perez P, Cortes J, Roca O, Masclans JR, Rello J. Effect of high-flow nasal cannula and body position on end-expiratory lung volume: a cohort study using electrical impedance tomography. Respir Care. 2013;58(4):589–96.
Corley A, Bull T, Spooner AJ, Barnett AG, Fraser JF. Direct extubation onto high-flow nasal cannulae post-cardiac surgery versus standard treatment in patients with a BMI ≥ 30: a randomised controlled trial. Intensive Care Med. 2015;41(5):887–94.
Maggiore SM, Idone FA, Vaschetto R, et al. Nasal high-flow versus Venturi mask oxygen therapy after extubation. Effects on oxygenation, comfort, and clinical outcome. Am J Respir Crit Care Med. 2014;190(3):282–8.
Vourc'h M, Asfar P, Volteau C, et al. High-flow nasal cannula oxygen during endotracheal intubation in hypoxemic patients: a randomized controlled clinical trial. Intensive Care Med. 2015;2015:14.
Kang BJ, Koh Y, Lim CM, et al. Failure of high-flow nasal cannula therapy may delay intubation and increase mortality. Intensive Care Med. 2015;41(4):623–32.
Messika J, Ben Ahmed K, Gaudry S, et al. Use of high-flow nasal cannula oxygen therapy in subjects with ARDS: a 1-year observational study. Respir Care. 2015;60(2):162–9.
Parke R, McGuinness S, Dixon R, Jull A. Open-label, phase II study of routine high-flow nasal oxygen therapy in cardiac surgical patients. Br J Anaesth. 2013;111(6):925–31.
Lucangelo U, Vassallo FG, Marras E, et al. High-flow nasal interface improves oxygenation in patients undergoing bronchoscopy. Crit Care Res Pract. 2012;2012(506382):506382.
Simon M, Braune S, Frings D, Wiontzek AK, Klose H, Kluge S. High-flow nasal cannula oxygen versus non-invasive ventilation in patients with acute hypoxaemic respiratory failure undergoing flexible bronchoscopy—a prospective randomised trial. Crit Care. 2014;18(6):712.
Parke RL, McGuinness SP. Pressures delivered by nasal high flow oxygen during all phases of the respiratory cycle. Respir Care. 2013;58(10):1621–4.
Roca O, Riera J, Torres F, Masclans JR. High-flow oxygen therapy in acute respiratory failure. Respir Care. 2010;55(4):408–13.
Rello J, Perez M, Roca O, et al. High-flow nasal therapy in adults with severe acute respiratory infection: a cohort study in patients with 2009 influenza A/H1N1v. J Crit Care. 2012;27(5):434–9.
Nagata K, Morimoto T, Fujimoto D, et al. Efficacy of high-flow nasal cannula therapy in acute hypoxemic respiratory failure: decreased use of mechanical ventilation. Respir Care. 2015;2015(23):04026.
Lenglet H, Sztrymf B, Leroy C, Brun P, Dreyfuss D, Ricard JD. Humidified high flow nasal oxygen during respiratory failure in the emergency department: feasibility and efficacy. Respir Care. 2013;57(11):1873–8.
Rittayamai N, Tscheikuna J, Praphruetkit N, Kijpinyochai S. Use of high-flow nasal cannula for acute dyspnea and hypoxemia in the emergency department. Respir Care. 2015;2015(9):03837.
Futier E, Paugam-Burtz C, Constantin JM, Pereira B, Jaber S. The OPERA trial - comparison of early nasal high flow oxygen therapy with standard care for prevention of postoperative hypoxemia after abdominal surgery: study protocol for a multicenter randomized controlled trial. Trials. 2013;14(341):341.
Miguel-Montanes R, Hajage D, Messika J, et al. Use of high-flow nasal cannula oxygen therapy to prevent desaturation during tracheal intubation of intensive care patients with mild-to-moderate hypoxemia. Crit Care Med. 2015;43(3):574–83.
Tiruvoipati R, Lewis D, Haji K, Botha J. High-flow nasal oxygen vs high-flow face mask: a randomized crossover trial in extubated patients. J Crit Care. 2010;25(3):463–8.
Brotfain E, Zlotnik A, Schwartz A, et al. Comparison of the effectiveness of high flow nasal oxygen cannula vs. standard non-rebreather oxygen face mask in post-extubation intensive care unit patients. Isr Med Assoc J. 2014;16(11):718–22.
Lee HY, Rhee CK, Lee JW. Feasibility of high-flow nasal cannula oxygen therapy for acute respiratory failure in patients with hematologic malignancies: a retrospective single-center study. J Crit Care. 2015;30(4):773–7.
Epstein AS, Hartridge-Lambert SK, Ramaker JS, Voigt LP, Portlock CS. Humidified high-flow nasal oxygen utilization in patients with cancer at Memorial Sloan-Kettering Cancer Center. J Palliat Med. 2011;14(7):835–9.
Hui D, Morgado M, Chisholm G, et al. High-flow oxygen and bilevel positive airway pressure for persistent dyspnea in patients with advanced cancer: a phase II randomized trial. J Pain Symptom Manag. 2013;46(4):463–73.
Roca O, de Acilu MG, Caralt B, Sacanell J, Masclans JR. Humidified high flow nasal cannula supportive therapy improves outcomes in lung transplant recipients readmitted to the intensive care unit because of acute respiratory failure. Transplantation. 2015;99(5):1092–8.
Peters SG, Holets SR, Gay PC. High-flow nasal cannula therapy in do-not-intubate patients with hypoxemic respiratory distress. Respir Care. 2013;58(4):597–600.
Mokart D, Geay C, Chow-Chine L, et al. High-flow oxygen therapy in cancer patients with acute respiratory failure. Intensive Care Med. 2015;2015:4.
Lemiale V, Resche-Rigon M, Azoulay E. Early non-invasive ventilation for acute respiratory failure in immunocompromised patients (IVNIctus): study protocol for a multicenter randomized controlled trial. Trials. 2015;15(372):372.
Kaji AH, Lewis RJ. Noninferiority trials: is a New Treatment almost as effective as another? JAMA. 2015;313(23):2371–2.
Ferrer M, Valencia M, Nicolas JM, Bernadich O, Badia JR, Torres A. Early NIV averts extubation failure in patients at risk trial. Am J Respir Crit Care Med. 2006;173(2):164–70.
Coudroy R, Jamet A, Petua P, Robert R, Frat JP, Thille A. High-flow nasal cannula oxygen therapy versus noninvasive ventilation in immunocompromised patients with acute respiratory failure: an observational cohort study. Ann Intensive Care. 2016;6(1):45.
Frat JP, Ragot S, Girault C, Perbet S, Prat G, Boulain T, Demoule A, Ricard JD, Coudroy R, Robert R, Mercat A, Brochard L, Thille AW. REVA network. Effect of non-invasive oxygenation strategies in immunocompromised patients with severe acute respiratory failure: a post-hoc analysis of a randomised trial. Lancet Respir Med. 2016;4(8):646–52.
Azoulay E, Pickkers P, Soares M, Perner A, Rello J, Bauer PR, van de Louw A, Hemelaar P, Lemiale V, Taccone FS, Martin Loeches I, Meyhoff TS, Salluh J, Schellongowski P, Rusinova K, Terzi N, Mehta S, Antonelli M, Kouatchet A, Barratt-Due A, Valkonen M, Landburg PP, Bruneel F, Bukan RB, Pène F, Metaxa V, Moreau AS, Souppart V, Burghi G, Girault C, Uva S, Montini L, Barbier F, Nielsen LB, Gaborit B, Mokart D, Chevret S, Efraim investigators and the Nine-I study group. Acute hypoxemic respiratory failure in immunocompromised patients: the Efraim multinational prospective cohort study. Intensive Care Med. 2017;43(12):1808–19.
Garner JS, Jarvis WR, Emori TG, Horan TC, Hughes JM. CDC definitions for nosocomial infections, 1988. Am J Infect Control. 1988;16(3):128–40.
Acknowledgements
Fisher & Payckle provided the high-flow oxygen devices to participating centers as to increase their ability to recruit several patients at the same time. None of the people listed in the author’s group has received any honorarium or fees for participation to this study.
Funding
The study has received a grant from the French Ministry of Health.
Availability of data and materials
All the data collected for this study are in the hands of Sylvie Chevret MD, PhD who is the methodologist of the trial and statistician for the study. All data will be available upon request.
Author information
Authors and Affiliations
Contributions
EA, VL, DM, and AD have drafted the initial version of the protocol and have requested funding from the Ministry of Health. SC has designed the study and planned the statistics. She also ran the interim analyses. SN, LA, FP, LK, and FB participated in study conception and to address initial discussions that helped obtain the grant. EA, VL, DM, AD, SN, LA, FP, LK FB. KK, FB, JR, AS, GL, JMC, JM, FW, AK, VP, PP, CG, SJ, JO, MY, NT, LB, CL, AL, NB, JHR, LP, AR, and MD also gave feedback on study design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript. All authors attended the investigators meeting, are responsible for all decisions regarding the study, are responsible for recruiting patients, collecting data and completing information on the eCRF.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the IRB of the St. Louis Hospital. All patients or relatives provided signed informed consent.
Consent for publication
All authors consent to see this protocol article published. All have given input on the submitted version and approved it.
Competing interests
None of the authors has any conflict of interest in relation with this study. The institutions of Elie Azoulay, Samir Jaber, Alexandre Demoule and Virginie Lemiale have received scientific support from Fisher & Payckle outside this study.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
SPIRIT 2013 Checklist: recommended items to address in a clinical trial protocol and related documents*. (PDF 142 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Azoulay, E., Lemiale, V., Mokart, D. et al. High-flow nasal oxygen vs. standard oxygen therapy in immunocompromised patients with acute respiratory failure: study protocol for a randomized controlled trial. Trials 19, 157 (2018). https://doi.org/10.1186/s13063-018-2492-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-018-2492-z