
Abstract
The REPIC (RNA EPItranscriptome Collection) database records about 10 million peaks called from publicly available m6A-seq and MeRIP-seq data using our unified pipeline. These data were collected from 672 samples of 49 studies, covering 61 cell lines or tissues in 11 organisms. REPIC allows users to query N6-methyladenosine (m6A) modification sites by specific cell lines or tissue types. In addition, it integrates m6A/MeRIP-seq data with 1418 histone ChIP-seq and 118 DNase-seq data tracks from the ENCODE project in a modern genome browser to present a comprehensive atlas of m6A methylation sites, histone modification sites, and chromatin accessibility regions. REPIC is accessible at https://repicmod.uchicago.edu/repic.
Similar content being viewed by others


Background
Over 150 chemical modifications have been identified in messenger RNAs (mRNAs) and non-coding RNAs (ncRNAs) [1]. Among them, N6-methyladenosine (m6A) is characterized as the most abundant and reversible mRNA internal modification [2, 3]. Numerous studies have emerged to establish m6A as a critical regulator of post-transcriptional gene expression programs which is involved with many cellular activities including splicing [4], translation efficiency [5], stability [6], export, and cytoplasmic localization [7] of m6A-modified mRNAs. Furthermore, m6A also impacts a series of physiological processes including, but not limited to, proliferation [8], development [9], neurogenesis [10], circadian rhythm [11], and embryonic stem cell differentiation [12].
With the advent of next-generation sequencing (NGS) technologies, several high-throughput sequencing methods (m6A-seq or MeRIP-seq [13, 14], PA-m6A-seq [15], m6A-LAIC-seq [16], miCLIP [17, 18], m6A-REF-seq [19], MAZTER-seq [20], and DART-seq [21]) have been developed to explore m6A modifications quantitatively across the entire transcriptome, paving the way for understanding their biological functions. These methods, especially m6A/MeRIP-seq, have been widely adopted to profile the m6A marks in a variety of cell lines and tissue types from multiple species. To better explore m6A data sets with increasing complexity, several databases (RMBase v2.0 [22], MeT-DB v2.0 [23], CVm6A [24]) and web servers (RNAmod [25], WHISTLE [26], SRAMP [27]) have been constructed to organize and integrate existing resources. Among these, RMBase v2.0 integrates information on sites of five or more types of RNA modifications, RBP binding sites, and single nucleotide polymorphisms, whereas MeT-DB v2.0 and CVm6A publish m6A peaks processed by their own pipelines from raw m6A sequencing data (Table 1). However, these databases have limitations. It has been shown that distinct m6A patterns occur in different developmental stages or tissue types, implying their dynamic regulation in a tissue-dependent manner [28]. Unfortunately, all of the above databases, except for CVm6A, simply combine m6A peaks across data sets without considering cell type or tissue specificity (Table 1). Furthermore, recent studies have uncovered associations between m6A modifications and promoters [29,30,31] or histone marks [32, 33], offering new insights into potential regulatory pathways and underlying mechanisms, through which m6A could influence transcriptional regulation and gene expression. However, to our knowledge, m6A modifications and epigenomic data have not been curated together well. New bioinformatic tools are needed for processing, analyzing, and visualizing the integration of these data.
Here, we present the REPIC (RNA EPItranscriptome Collection) database, which currently focuses on integrating m6A modifications with ENCODE epigenomic data (Table 1). The m6A modification peaks are generated by re-processing publicly available m6A-seq and MeRIP-seq data sets using a unified customized pipeline. REPIC allows users to query m6A modification sites by cell lines or tissue types with a user-friendly interface and provides a built-in genome browser for visualization. Overall, REPIC is a new resource designed to allow users to explore cell/tissue-specific m6A modifications and investigate potential interactions between m6A modifications and histone marks or chromatin accessibility.
Construction and content
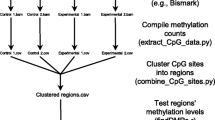
The REPIC database collected m6A modifications and epigenomic sequencing data from different species. We designed a modern, user-friendly web portal for querying m6A modification sites and an interactive genome browser empowered by GIVE [34] for data visualization (Fig. 1a). The web application of the REPIC database was constructed using Apache v2.4.18, MySQL v5.7.25, and PHP v7.2.14. The data processing procedures starting from raw data sources are shown in Fig. 1b. To better disseminate the resource and facilitate downstream analysis, we provide curated data that can be downloaded from the REPIC database website.

a Overall design of the REPIC database. b Schema of the customized pipeline for m6A-seq or MeRIP-seq data processing
High-throughput sequencing data
Raw m6A-seq and MeRIP-seq data were manually collected through an extensive literature search and then retrieved from the Gene Expression Omnibus (GEO) and the Sequence Read Archive (SRA). In total, 607 m6A-seq and 544 MeRIP-seq run data were obtained from SRA. After merging different runs in the same experiment and excluding unpaired input-IP samples, 672 samples—which consisted of 339 pairs of input-IP data from 49 studies, covering 61 cell lines or tissue types in 11 organisms—were used for database construction (Additional file 1: Table S1). For epigenomic data, a total of 118 DNase-seq peak sets from 29 cell lines or tissue types, and 1418 histone ChIP-seq peak sets from 27 histone marks in 22 cell lines or tissue types in human and mouse, matching with curated m6A modification data, were downloaded from the ENCODE website (Additional file 1: Table S2 and S3).
Genome annotation data
Human and mouse genome sequences and gene annotations were acquired from the UCSC Genome Browser [35] and GENCODE [36], respectively. Arabidopsis thaliana genome sequences and gene annotations were obtained from the Arabidopsis Information Resource (TAIR) [37]. The rest were downloaded from the Ensembl website [38]. The widespread versions of genome sequences and gene annotations for each of the 11 organisms were chosen for further analysis (Additional file 1: Table S4).
Raw m6A-seq and MeRIP-seq data reprocessing
The aforementioned 339 pairs of input-IP data were re-processed by our customized pipeline [39, 40] (Fig. 1b). Briefly, adapters of raw sequencing data were clipped away by Cutadapt v1.15 [41]. Reads longer than 15 nt after trimming were first mapped to ribosomal RNAs (rRNAs) by HISAT2 v2.1.0 [42]. All unmapped reads were then aligned to genomes using HISAT2 v2.1.0 with default parameters. For samples with low mapping ratios, we used FastQ Screen [43] to find possible contaminants in those sample reads. To check library complexity, PCR duplicates were evaluated by MarkDuplicates from Picard v2.17.10 [44]. We then calculated the PCR duplicate proportion (PDP), which we defined as the number of PCR duplicate reads divided by the total number of mapped reads. Another three metrics, non-redundant fraction (NRF) and PCR bottlenecking coefficients 1 (PBC1) and 2 (PBC2), were quantified using ENCODE standards [45]. Input samples from m6A-seq and MeRIP-seq data were used to estimate gene expression levels by StringTie v1.3.4d [46]. If the library type was strand-specific, we further divided the sequence alignment data by strands. For visualization, log2 fold enrichment levels of m6A were calculated using bamCompare, and gene expression levels were reported in bins per million mapped reads (BPM) using bamCoverage from deepTools v3.0.2 [47]. exomePeak [48], MeTPeak [49], and MACS2 v2.1.1 [50] were used to detect peaks. For exomePeak and MeTPeak, parameters were set as follows: PEAK_CUTOFF_FDR = 0.05, WINDOW_WIDTH = 50, SLIDING_STEP = 10, MINIMAL_MAPQ = 20, FOLD_ENRICHMENT = 2, and REMOVE_LOCAL_TAG_ANOMALITIES=F. The values of the parameters FRAGMENT_LENGTH and READ_LENGTH varied under different library settings. Parameters in MACS2 were set as follows: -f BAM -B --SPMR --nomodel --keep-dup all. The values of the options -g, --tsize, and --extsize varied under different library settings. Finally, HOMER v4.9 [51] was used for motif enrichment analysis based on the top 2000 peaks ranked by their fold enrichment levels.
Utility and discussion
Evaluation of m6A-seq and MeRIP-seq data quality
We applied our pipeline to re-process all collected m6A-seq and MeRIP-seq samples. As rRNAs could potentially interfere with mRNA expression quantification and peak calling, we first interrogated the rRNA content in each sample. rRNA reads comprised less than 30% of total reads in 566 samples (85.0% of the total), while 371 samples (55.7% of the total) contained a proportion of rRNA reads below 5% (Fig. 2a), suggesting that most samples were not subject to rRNA contamination. Next, we examined the counts of reads mapped to the genomes after filtering out rRNA reads. Five hundred seventy-one samples (85.7%) were shown to be of high quality with a genome mapping ratio greater than 75% (Fig. 2b). Sixteen human and 22 mouse samples with a low genome mapping ratio (< 60%) were detected as containing viral infection, vector or mycoplasma contamination, or other unknown conditions.

The quality of m6A-seq or MeRIP-seq reads mapping. Boxplots depicting the distribution of reads mapped to a rRNAs and b genomes in the input and IP samples, respectively. The y-axis in a and b represents the percentage of reads mapped to rRNAs and non-rRNA reads mapped to genomes, respectively. Both left-side panels show the whole range of the ratios and the right-side panels of a and b zoom in on the ranges of 0–5% and 75–100%, respectively
To further evaluate data quality, we assessed the library complexity of all samples by four metrics: PDP, NRF, PBC1, and PBC2, with the last three as defined by the ENCODE project [45]. The PDP values indicated that around 75% of the samples contained PCR duplicate proportions of greater than 50% (Additional file 2: Figure S1A), whereas the NRF values showed that only about 25% of the samples had a fraction of distinct, uniquely mapping reads greater than 50% (Additional file 2: Figure S1B). Both PDP and NRF values across the samples implied that multiple reads in the same positions of the genomes were prevalent. However, the decision of whether to remove them as PCR duplicates is an open question, since it is difficult to distinguish between artifacts of PCR amplification and real transcriptional events using current computational methods. Furthermore, direct removal of duplicate reads with the same mapping coordinates may introduce unwanted bias [52, 53]. Therefore, our pipeline keeps duplicated reads for downstream analysis. Unlike for PDP and NRF, about 90% of the input samples and 75% of the IP samples showed no severe (PBC1 > 0.5) or moderate (PBC2 > 3) levels of PCR bottlenecking (Additional file 2: Figure S1C and S1D) according to ENCODE standards. Overall, the two metrics PBC1 and PBC2 indicated that the library complexity of the majority of samples was of acceptable quality; thus, we considered them for further analysis. Nevertheless, we note that some characteristics of RNA biogenesis are more complicated than DNAs, so new metrics may need to be developed for the evaluation of RNA library complexity.
Three peak calling tools—exomePeak, MeTPeak, and MACS2—have been widely used for m6A peak detection. exomePeak and MeTPeak were developed by the same group, but their algorithms vary. MeTPeak outperforms exomePeak based on robustness against data variance and detection of lowly enriched peaks [49]. However, with our processed data sets, exomePeak achieves better motif enrichment than MeTPeak. Unlike exomePeak and MeTPeak, both of which, by design, detect peaks across the transcriptome, MACS2 determines peaks genome wide. Thus, we can use MACS2 to obtain intronic and intergenic peaks. Because the algorithms of all three tools each have unique advantages, we applied them all to identify m6A peaks from the collected samples using fixed parameters. To assess the similarity of the peak sets identified by different tools, we adopted the Jaccard Index (JI) and Simpson Index (SI). JI is defined as the number of intersecting bases between two peak sets divided by the number of bases in the union of the two peak sets [54], and SI measures the ratio of the number of intersecting bases between two peak sets to the number of bases in the smaller of the two peak sets [55]. Thus, by definition, a given pair of peak sets has a higher SI than JI; the indexes have the same numerator, but the SI has a smaller denominator. To limit the comparisons at the transcriptome level, we considered only MACS2 peaks that overlapped with annotated transcripts. Unexpectedly, only about 13.6% and 3.0% of the peak sets from MACS2 had 50% or greater complete overlap (JI > 0.5) with those from exomePeak and MeTPeak, respectively (Fig. 3a, b). This observation indicated poor reproducibility between peak sets called by MACS2 and those by exomePeak or MeTPeak for the same given data sets. On the contrary, about 77.4% of the peak sets from exomePeak have JI > 0.5 when compared with those from MeTPeak (Fig. 3c). In addition, about 73.0% and 86.6% of the peak sets from exomePeak have SI > 0.75 with those from MACS2 and MeTPeak, respectively (Fig. 3a, c). However, the proportion of the peak sets between MACS2 and MeTPeak with the same SI was reduced to 37.7% (Fig. 3b). It suggests that peaks called by MACS2 and MeTPeak achieve lower consistency than those called by MACS2 and exomePeak. Taken together, exomePeak and MeTPeak agreed on over 75% of peak sets (JI > 0.5 or SI > 0.75), while MACS2 recovered limited peaks from exomePeak and especially MeTPeak.

Evaluation of similarities of peak sets generated by three peak calling tools. Scatter plots showing the distributions of the Jaccard Index and Simpson Index from comparisons of a exomePeak versus MACS2, b MeTPeak versus MACS2, and c exomePeak versus MeTPeak across all samples. Paired-end and single-end sequencing types are represented by triangles and circles, respectively. Species are indicated by colors
Cell- or tissue-specific m6A modifications
As genes are expressed in a tissue-specific manner, we asked whether m6A modifications possess similar characteristics. According to the metagene profiles of m6A in mRNAs [56], we first considered five distinct genomic features: 5′ UTR, CDS, stop codon regions (± 200 bp around the stop codons), 3′ UTR, and whole regions. We then examined the top 2000 genes ranked by coefficients of variation (CV) of fold enrichment levels of m6A peaks at these regions across human cell lines and tissues. By comparing the m6A peak enrichment between samples at the 5′ UTR (Additional file 2: Figure S2A), CDS (Additional file 2: Figure S2B), 3′ UTR (Additional file 2: Figure S2C), and whole regions (Additional file 2: Figure S2D), we observed the strongest correlations among samples from the same cell lines or tissue types at stop codon regions (Fig. 4a), even when they were collected from different studies or labs. This phenomenon was also presented in the t-distributed stochastic neighbor embedding (t-SNE) [57] plot; samples from the same cell or tissue type were clustered together and clearly separated from other distinct groups (Fig. 4b). These results suggest that some highly dynamic m6A modifications at stop codon regions more so than those at other functional regions tend to be tightly controlled, perhaps in order to regulate cellular activities and processes in a cell line- or tissue type-specific manner, in response to different physiological stimuli or conditions.

Cell- or tissue-specific m6A modifications. a Heatmap depicting the Pearson correlation of different human cell lines and tissues of the top 2000 genes ranked by CVs of fold enrichment levels of m6A peaks at stop codon regions (± 200 bp around the stop codons). The dendrogram was constructed using complete linkage based on Euclidean distances. Each row label represents the sample information in the format of “input_IP”. bt-SNE plot displaying grouping patterns of different cell/tissue samples in a lower-dimensional space for the same data in a. Each dot represents a sample. Cell/tissue types are indicated by colors
To offer insight into the cell line or tissue specificity of m6A modifications, REPIC supports the query of m6A modifications by cell lines or tissue types (Fig. 5a). On the Search page, we list options for all available cell lines and tissue types, next to filtering options that include the number of peak sites in the gene of interest and samples from which peaks were called (Fig. 5b). Once the submitted query is complete, a report will be presented in a user-friendly interface with the following information for each peak: genome position, other tools that identify an overlapping peak, fold enrichment, and genomic feature annotation (Additional file 2: Figure S3A). More sample information can be found in a separate window, including the data source, read mapping statistics, metagene profiles, and results from motif enrichment analysis (Additional file 2: Figure S3B).

Screenshots of the web interfaces of the REPIC database. a The Home page. b The Search page. c Taking the query region near gene NANOG as an example, we show a visualization of m6A peaks, histone modifications, and chromatin accessibility in the genome browser
Visualization of m6A modifications and epigenomic data
The query on the Search page is limited to genes. To better display multi-dimensional m6A modification information across the entire genome, REPIC provides a genome browser empowered by GIVE to visualize m6A peaks, fold enrichment, and gene expression. As increasing evidence has shown that chromatin accessibility as well as epigenetic marks such as histone modifications defines the cell/tissue types [58, 59], we built REPIC to integrate DNase-seq and histone ChIP-seq data in order to investigate the possible correlations between these epigenomic characteristics and m6A modifications. As a result, a total of 3225 tracks comprising seven distinct track types (Additional file 1: Table S5) constitute the built-in genome browser. Like the UCSC Genome Browser or other similar genome browsers, a user can select multiple tracks to interactively display peak or expression profile data at a specific genomic location. In an example demonstrating the utility of the browser shown in Fig. 5c, we observe that H3K4me3 and DNase-seq peaks are located in the promoter region of the NANOG gene, indicating that it is actively transcribed in hESCs [12]. We also note that m6A modifications at the stop codon region are enriched with H3K36me3 peaks, which is consistent with the recently reported H3K36me3-dependent mechanism of m6A modification deposition [32].
Future directions
As m6A modification detection technology has been applied to a variety of cell/tissue types with different conditions in distinct species, we will continue to collect new m6A/MeRIP-seq samples. In addition, with the increasing availability of transcriptome-wide sequencing data of m6A modifications at a single-nucleotide resolution as well as other RNA modifications including m1A, m5C, m7G, Ψ, and Nm, we will expand REPIC to catalog those as well. Another future development will be the integration of non-epitranscriptomic data such as RBP binding sites, GWAS, and GTEx data [60] to facilitate assessment and interpretation of RNA modifications.
Conclusions
The current release of the REPIC database integrates millions of m6A peaks called by three popular tools from various cell/tissue types of multiple species. REPIC allows users to query m6A modification sites by specific cell lines or tissue types. Furthermore, hundreds of epigenomic data sets including chromatin accessibility and histone marks are included with the built-in genome browser to facilitate the interpretation of the functions of certain cell/tissue-specific m6A modifications, revealing their direct or indirect roles in influencing chromatin states and transcriptional regulation.
Availability of data and materials
The lists of public m6A/MeRIP-seq, histone ChIP-seq, and DNase-seq data sets are also available in Additional file 1.
Our customized pipeline is freely available on GitHub (https://github.com/shunliubio/easym6A) [39] and Zenodo (https://doi.org/10.5281/zenodo.3742549) [40] under the GNU General Public License (GPL-v3.0).
All 339 m6A peak sets can be downloaded from the REPIC data download center [61].
References
Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK, de Crecy-Lagard V, Ross R, Limbach PA, Kotter A, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46:D303–7.
Yang Y, Hsu PJ, Chen YS, Yang YG. Dynamic transcriptomic m6A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28:616–24.
Shi H, Wei J, He C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell. 2019;74:640–50.
Louloupi A, Ntini E, Conrad T, Orom UAV. Transient N-6-methyladenosine transcriptome sequencing reveals a regulatory role of m6A in splicing efficiency. Cell Rep. 2018;23:3429–37.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C. N6-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–99.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–20.
Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, Sha J, Huang X, Guerrero L, Xie P, et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. Elife. 2017;6:e31311.
Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, Tienda SM, Chryplewicz A, Zhu AC, Yang Y, et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018;20:1074–83.
Zhao BS, Wang X, Beadell AV, Lu Z, Shi H, Kuuspalu A, Ho RK, He C. m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature. 2017;542:475–8.
Yoon KJ, Ringeling FR, Vissers C, Jacob F, Pokrass M, Jimenez-Cyrus D, Su Y, Kim NS, Zhu Y, Zheng L, et al. Temporal control of mammalian cortical neurogenesis by m6A methylation. Cell. 2017;171:877–89 e817.
Fustin JM, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M, Isagawa T, Morioka MS, Kakeya H, Manabe I, Okamura H. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell. 2013;155:793–806.
Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15:707–19.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–6.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–46.
Chen K, Lu Z, Wang X, Fu Y, Luo GZ, Liu N, Han D, Dominissini D, Dai Q, Pan T, He C. High-resolution N6-methyladenosine (m6A) map using photo-crosslinking-assisted m6A sequencing. Angew Chem Int Ed Engl. 2015;54:1587–90.
Molinie B, Wang J, Lim KS, Hillebrand R, Lu ZX, Van Wittenberghe N, Howard BD, Daneshvar K, Mullen AC, Dedon P, et al. m6A-LAIC-seq reveals the census and complexity of the m6A epitranscriptome. Nat Methods. 2016;13:692–8.
Ke S, Alemu EA, Mertens C, Gantman EC, Fak JJ, Mele A, Haripal B, Zucker-Scharff I, Moore MJ, Park CY, et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 2015;29:2037–53.
Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12:767–72.
Zhang Z, Chen LQ, Zhao YL, Yang CG, Roundtree IA, Zhang Z, Ren J, Xie W, He C, Luo GZ. Single-base mapping of m6A by an antibody-independent method. Sci Adv. 2019;5:eaax0250.
Garcia-Campos MA, Edelheit S, Toth U, Safra M, Shachar R, Viukov S, Winkler R, Nir R, Lasman L, Brandis A, et al. Deciphering the “m6A code” via antibody-independent quantitative profiling. Cell. 2019;178:731–47 e716.
Meyer KD. DART-seq: an antibody-free method for global m6A detection. Nat Methods. 2019;16:1275–80.
Xuan JJ, Sun WJ, Lin PH, Zhou KR, Liu S, Zheng LL, Qu LH, Yang JH. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018;46:D327–34.
Liu H, Wang H, Wei Z, Zhang S, Hua G, Zhang SW, Zhang L, Gao SJ, Meng J, Chen X, Huang Y. MeT-DB V2.0: elucidating context-specific functions of N6-methyl-adenosine methyltranscriptome. Nucleic Acids Res. 2018;46:D281–7.
Han Y, Feng J, Xia L, Dong X, Zhang X, Zhang S, Miao Y, Xu Q, Xiao S, Zuo Z, et al. CVm6A: a visualization and exploration database for m6As in cell lines. Cells. 2019;8:168.
Liu Q, Gregory RI. RNAmod: an integrated system for the annotation of mRNA modifications. Nucleic Acids Res. 2019;47:W548–55.
Chen K, Wei Z, Zhang Q, Wu X, Rong R, Lu Z, Su J, de Magalhaes JP, Rigden DJ, Meng J. WHISTLE: a high-accuracy map of the human N6-methyladenosine (m6A) epitranscriptome predicted using a machine learning approach. Nucleic Acids Res. 2019;47:e41.
Zhou Y, Zeng P, Li YH, Zhang Z, Cui Q. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016;44:e91.
Yue Y, Liu J, He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015;29:1343–55.
Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC, Aspris D, Migliori V, Bannister AJ, Han N, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature. 2017;552:126–31.
Bertero A, Brown S, Madrigal P, Osnato A, Ortmann D, Yiangou L, Kadiwala J, Hubner NC, de Los Mozos IR, Sadee C, et al. The SMAD2/3 interactome reveals that TGFβ controls m6A mRNA methylation in pluripotency. Nature. 2018;555:256–9.
Slobodin B, Han R, Calderone V, Vrielink J, Loayza-Puch F, Elkon R, Agami R. Transcription impacts the efficiency of mRNA translation via co-transcriptional N6-adenosine methylation. Cell. 2017;169:326–37 e312.
Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, Chen Z, Deng X, Xiao G, Auer F, et al. Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature. 2019;567:414–9.
Wang Y, Li Y, Yue M, Wang J, Kumar S, Wechsler-Reya RJ, Zhang Z, Ogawa Y, Kellis M, Duester G, Zhao JC. N6-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat Neurosci. 2018;21:195–206.
Cao X, Yan Z, Wu Q, Zheng A, Zhong S. GIVE: portable genome browsers for personal websites. Genome Biol. 2018;19:92.
Haeussler M, Zweig AS, Tyner C, Speir ML, Rosenbloom KR, Raney BJ, Lee CM, Lee BT, Hinrichs AS, Gonzalez JN, et al. The UCSC Genome Browser database: 2019 update. Nucleic Acids Res. 2019;47:D853–8.
Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766–73.
Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, Sasidharan R, Muller R, Dreher K, Alexander DL, Garcia-Hernandez M, et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 2012;40:D1202–10.
Cunningham F, Achuthan P, Akanni W, Allen J, Amode MR, Armean IM, Bennett R, Bhai J, Billis K, Boddu S, et al. Ensembl 2019. Nucleic Acids Res. 2019;47:D745–51.
Liu S. easym6A: process m6A/MeRIP-seq data in a single or batch job mode. Github. http://www.github.com/shunliubio/easym6A(2020). Accessed 7 April 2020.
Liu S. easym6A: process m6A/MeRIP-seq data in a single or batch job mode. Zenodo. 2020. https://doi.org/10.5281/zenodo.3742549.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:3.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60.
Wingett SW, Andrews S. FastQ Screen: a tool for multi-genome mapping and quality control. F1000Res. 2018;7:1338.
The Picard toolkit: http://broadinstitute.github.io/picard/. Accessed 19 Feb 2018.
Library complexity of ENCODE standards: https://www.encodeproject.org/data-standards/terms/#library. Accessed 7 April 2020.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–5.
Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42:W187–91.
Meng J, Cui X, Rao MK, Chen Y, Huang Y. Exome-based analysis for RNA epigenome sequencing data. Bioinformatics. 2013;29:1565–7.
Cui X, Meng J, Zhang S, Chen Y, Huang Y. A novel algorithm for calling mRNA m6A peaks by modeling biological variances in MeRIP-seq data. Bioinformatics. 2016;32:i378–85.
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137.
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89.
Fu Y, Wu PH, Beane T, Zamore PD, Weng Z. Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers. BMC Genomics. 2018;19:531.
Parekh S, Ziegenhain C, Vieth B, Enard W, Hellmann I. The impact of amplification on differential expression analyses by RNA-seq. Sci Rep. 2016;6:25533.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2.
Fuxman Bass JI, Diallo A, Nelson J, Soto JM, Myers CL, Walhout AJ. Using networks to measure similarity between genes: association index selection. Nat Methods. 2013;10:1169–76.
Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet. 2014;15:293–306.
van der Maaten L, Hinton G. Visualizing data using t-SNE. J Mach Learn Res. 2008;9:2579–605.
Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, Sheffield NC, Graf S, Huss M, Keefe D, et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011;21:1757–67.
Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18.
Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, et al. Human genomics. The human transcriptome across tissues and individuals. Science. 2015;348:660–5.
The REPIC data download center: https://repicmod.uchicago.edu/repic/download.php. Accessed 7 April 2020.
Peer review information
Yixin Yao was the primary editor of this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Review history
The review history is available as Additional file 3.
Funding
MC is supported by R01 GM126553 and a Sloan Foundation Research Fellowship. CH is supported by HG008935.
Author information
Authors and Affiliations
Contributions
MC and SL planned and designed the project in consultation with CH. SL performed the analyses and built the database and its web interface. MC, SL, and AZ wrote the manuscript. All the authors have reviewed, commented, and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval is not applicable to this study.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
The list of sample information for the 339 input-IP paired samples. Table S2. The data set list of histone ChIP-seq peaks from ENCODE. Table S3. The data set list of DNase-seq peaks from ENCODE. Table S4. The genome assembly versions and gene annotation sources of 11 organisms. Table S5. The descriptions of tracks in the genome browser.
Additional file 2: Figure S1.
Library complexity of m6A-seq or MeRIP-seq data. Figure S2. Correlation of m6A modifications in human cell lines and tissues categorized by genomic features. Figure S3. An example of the query of m6A modifications for a given gene.
Additional file 3.
Review history.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, S., Zhu, A., He, C. et al. REPIC: a database for exploring the N6-methyladenosine methylome. Genome Biol 21, 100 (2020). https://doi.org/10.1186/s13059-020-02012-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13059-020-02012-4