
Abstract
Background
Mutation rates vary across the genome. Many trans factors that influence mutation rates have been identified, as have specific sequence motifs at the 1–7-bp scale, but cis elements remain poorly characterized. The lack of understanding regarding why different sequences have different mutation rates hampers our ability to identify positive selection in evolution and to identify driver mutations in tumorigenesis.
Results
Here, we use a combination of synthetic genes and sequences of thousands of isolated yeast colonies to show that intrinsic DNA curvature is a major cis determinant of mutation rate. Mutation rate negatively correlates with DNA curvature within genes, and a 10% decrease in curvature results in a 70% increase in mutation rate. Consistently, both yeast and humans accumulate mutations in regions with small curvature. We further show that this effect is due to differences in the intrinsic mutation rate, likely due to differences in mutagen sensitivity and not due to differences in the local activity of DNA repair.
Conclusions
Our study establishes a framework for understanding the cis properties of DNA sequence in modulating the local mutation rate and identifies a novel causal source of non-uniform mutation rates across the genome.
Similar content being viewed by others


Background
Mutation is the ultimate source of genetic diversity. Therefore, the measurement of mutation rate and, particularly, the identification of the trans factors and cis elements that influence mutation rate are a focus of intense interest in evolutionary biology. A large number of trans factors influencing mutation rate have been identified [1], such as chromatin remodelers, histone-modifying enzymes, and other DNA-binding proteins [2,3,4]. In addition, replication timing [5,6,7,8,9] and transcription rate [10,11,12,13,14] also affect mutation rate.
Cis elements may play a more important role in determining the local mutation rate, yet remain poorly understood. Studies of cis elements that determine local mutation rate have been limited to the scale of a few neighboring nucleotides around a mutation site for the past few decades [15,16,17,18].
There is comprehensive cis information in the shape of DNA. Although the double-helix structure of DNA is usually described as a twisted ladder, the steps of the ladder are not rigidly aligned. The local shape of DNA is affected by the interactions of neighboring bases [19, 20]. For example, the depth and width of the minor and major grooves vary depending on the local sequence. Such variation in DNA shape affects the ability of proteins to bind to DNA and the accessibility of each nucleotide [20, 21] and, therefore, is under purifying selection [22]. Through its effect on DNA-protein and/or DNA-solvent interactions, the shape of the double helix may influence the local mutation rate. However, the role of DNA shape in influencing local mutation rate has not been systematically studied. Here, we provided several lines of evidence that intrinsic DNA curvature affects the local mutation rate in a quantitative and predictable manner. Our study therefore expands our knowledge of cis elements that regulate mutation rate by integrating information regarding the physical shape of the double helix and develops a new framework to understand the evolution of local mutation rate.
Results and discussion
Characterization of the mutational landscape of URA3
To quantitatively determine how cis elements affect the local mutation rate, we first characterized the mutational landscape of an endogenous gene, URA3, in Saccharomyces cerevisiae. URA3 encodes an enzyme required for uracil synthesis and converts the non-toxic molecule 5-fluoroorotic acid (5-FOA) into the toxic 5-fluorouracil. Only cells bearing loss-of-function mutations in URA3 can survive on 5-FOA plates, making URA3 a model gene to study mutation rate [5, 23]. Here, we cultured wild-type yeast in synthetic complete (SC) media for 24 h to allow mutations to accumulate and spread these cells onto a 5-FOA plate (Fig. 1a). We then sequenced URA3 of each randomly picked visible colony and identified mutations. We performed 135 biological replicates in parallel and sequenced a total of ~ 1000 URA3 variants from 135 plates (Additional file 1: Table S1). Identical mutations (same type at the same position) identified on the same plate were counted only once because such mutations are most likely resulted from cell proliferation from a single mutation and not independent identical mutations.

The mutational landscape of URA3. a A schematic description of the experimental design. Mutations were accumulated in SC liquid medium and ura3 mutants were selected on 5-FOA plates. b Mutational landscape of all potential nonsense mutation sites, which were defined as sites where a point mutation can result in a stop codon. Each bar represents a potential nonsense mutation site. c The observed (red arrow) and expected (histogram showing 1000 permutations) standard deviation of the numbers of nonsense mutations on all potential nonsense mutation sites of URA3
To measure bias in mutation rate, we need to determine the number of observed mutations and to compare it with the number expected if the mutation rate was uniform. As the missense mutations that would permit growth on 5-FOA is unknown, we focused our analysis on nonsense mutants. There are 104 potential nonsense mutation sites in URA3. For each of them, we counted the number of 5-FOA plates where each nonsense mutation was observed (Fig. 1b). This number varied between 0 and 8 (Fig. 1b). To determine if this variation in frequency could be fully explained by the inherently stochastic nature of mutation, we randomly assigned each of the observed 154 nonsense mutations to a potential nonsense mutation site. We then calculated the standard deviation of the observed numbers of nonsense mutations on these sites and that in the permutation. The observed standard deviation was significantly greater than the random expectation (P < 0.001, Fig. 1c), suggesting the presence of cis elements that affect the local mutation rate.
A nonsense mutation may not always lead to a loss of function, especially when it occurs near the stop codon. This would also lead to a non-Poisson distribution of observed mutations. To exclude this confounding factor, we repeated the permutation test using only the first two thirds of the coding sequence. Again, the observed standard deviation was significantly greater than the random expectation (Additional file 1: Figure S1a). Similar results were also obtained when we performed the permutation test separately for the 54 nonsense transitions and the 100 nonsense transversions (Additional file 1: Figure S1b-c). Taken together, the variation in the frequency of nonsense mutations within URA3 suggests the presence of cis elements that modulate local mutation rate.
Mutations in URA3 tend to occur in DNA regions with a smaller intrinsic DNA curvature
One possible explanation for the non-Poisson distribution of observed nonsense mutations is the difference in the mutation rate into a stop codon of each of the four bases. Nucleotides A and T had a lower mutation rate than G and C (Additional file 1: Figure S2), likely explained by the AT-rich nature of the three stop codons. That is, G>A and C>T transitions often result in stop codons but A>G and T>C transitions do not. To explore the predictive power of the nucleotide at each position and to identify additional cis sequence features predictive of local mutation rates, we constructed a set of linear models that take into account various sequence features (Table 1). Including the nucleotide at the potential nonsense site in the linear model decreases the Akaike information criterion (AIC) of the model, indicating an increase in the model’s ability to predict mutation rates (Table 1, model 1 and model 2). Surprisingly, including the + 1 and − 1 bases into the model did not further improve the predictive power nor did including the heptanucleotide sequence context (Table 1, models 3 and 4).
To identify additional DNA sequence features predictive of local mutation rates, we used a sliding window to divide the URA3 gene into overlapping regions of L nucleotides (L = 10, 20 …, or 100 bp). We calculated the average mutation rate in each region as the total number of observed nonsense mutations in this region normalized by the number of potential nonsense mutation sites (Additional file 1: Figure S3a). For each region, we then calculated 17 DNA properties such as GC content, thermodynamic characteristics, groove properties, and DNA shape features using well-established computational methods [19, 24] (Additional file 1: Figure S3b). Finally, for each window size, we calculated the correlation between mutation rate and each of the DNA properties.
Over a large range of window sizes, mutation rate was most strongly correlated with intrinsic DNA curvature, defined as the sequence-dependent deflection of DNA axis due to the interaction between neighboring base pairs [25] (e.g., for a window size L of 100 bp, ρ = − 0.49, P = 2 × 10− 5, Spearman’s correlation, Fig. 2a, b). Consistently, including intrinsic DNA curvature into the aforementioned linear model enhances its predictive power (Table 1, models 5 and 6). Adding the information of DNA curvature to the model only including the nucleotide at the potential nonsense site increases the adjusted coefficient of determination (r2) from 0.21 to 0.25, indicating that DNA curvature explains ~ 4% of the total variance in the per-base-pair mutation rate in URA3. It is worth noting that tilt, the DNA property exhibiting the second strongest correlation with mutation rate, is a component of intrinsic DNA curvature [25].

Intrinsic DNA curvature is negatively correlated with mutation rate. a Correlation between mutation rate and the value of each DNA feature in sliding windows (window length L). These features include GC content (orange), thermodynamic characteristics (purple), groove properties (green), intra- and inter-base pair DNA shape features (cyan), and integrated DNA shape features (blue). Intra- and inter-base pair DNA shape features are shown in cartoons, where a square represents a base and a rectangle represents a base pair. P values were calculated from the Spearman’s correlation. b, c Example scatter plots of URA3 (b) and of CAN1 (c). Each dot represents a region of length L (= 100 bp). d The average intrinsic DNA curvature of DNA regions surrounding the 882 observed mutation sites (red arrow) was significantly smaller than the random expectation (histogram showing 1000 permutations) in the yeast genome. P value was calculated with a permutation test
The correlation between mutation rate and DNA curvature was not confounded by GC content [17, 26] which in our data was not correlated with mutation rate (Fig. 2a). We previously showed that nucleosome binding suppresses spontaneous C>T transitions [27]. To quantitatively determine the relationship between mutation rate, nucleosome occupancy, and DNA curvature, we performed high-throughput sequencing on nucleosome-protected DNA fragments. The correlation between DNA curvature and mutation rate persisted after controlling for nucleosome occupancy (partial rURA3 = − 0.6, P = 1 × 10− 8), suggesting that the relationship between mutation rate and DNA curvature is not due to differences in nucleosome occupancy.
As a form of experimental cross-validation to determine if our results from URA3 are generalizable to other genes, we used an independently generated set of mutations in the yeast gene CAN1 [23], for which nonsense mutations were selected using the arginine analogue canavanine. Intrinsic DNA curvature is also predictive of mutation rate in CAN1 (Fig. 2c and Additional file 1: Table S2). In addition, nonsense mutations were reported to be unevenly distributed across sites within three human genes associated with Mendelian disease, MECP2, NF1, and RB1 [28], and within a tumor suppressor gene TP53 [29]. Consistently, intrinsic DNA curvature around a potential nonsense site was also negatively associated with the mutation rate of the site in each of these four genes (Additional file 1: Table S3).
Mutations in yeast and in humans accumulate in DNA regions with a smaller intrinsic DNA curvature
To determine if DNA curvature affects mutation rate at the genomic scale, we used a mutation accumulation assay in which spontaneous mutations accumulate at ~ 100× the normal rate due to a mutation in a gene related to DNA mismatch repair, MSH2 [30]. We retrieved all 882 mutations that were supported by an at least 20× coverage in the high-throughput sequencing data. We calculated the intrinsic DNA curvature of a region from 50 bp upstream to 50 bp downstream of each mutation. As a control, we randomly chose 882 sites with identical 3-nucleotide contexts (the mutation site, + 1, and − 1 sites) from the rest of the genome. We performed this random sampling procedure 1000 times. We found that the observed mutations were located in regions with a smaller intrinsic DNA curvature (P = 0.04, permutation test, Fig. 2d). It suggests that in the genome as a whole, regions with a smaller intrinsic DNA curvature have higher mutation rates.
Mutations generate genetic variation among cells within multi-cellular individuals, and somatic mutations play a vital role in cancer development and progression. Mutations in tumors are distributed unevenly across the genome and within individual genes [2, 3, 9, 16, 31]. We therefore performed the same genome-scale analysis as in yeast using 10,429 cancer samples from 26 cancer types collected in The Cancer Genome Atlas (TCGA) database [32]. We calculated the average intrinsic curvature of the DNA regions from 50 bp upstream to 50 bp downstream of each identified single nucleotide variant (SNV) for each cancer type. As a control, we randomly chose the same number of DNA sites from the genome. Consistent with the results in yeast, mutations were significantly enriched in regions with a smaller intrinsic DNA curvature in all cancer types (P < 0.001, permutation test, Fig. 3 and Additional file 1: Figure S4), suggesting that intrinsic DNA curvature reduces mutation rates in human tumor cells. To determine if the relationship between DNA curvature and mutation rate varies among genomic regions, we restricted SNVs in three well-annotated genomic regions, 5′ untranslated region (UTR), coding sequences, and 3′ UTR, and obtained the distribution of the expected DNA curvature by only sampling DNA sequences from the corresponding genomic regions. Intriguingly, DNA curvature was negatively associated with mutation rate only in coding sequences (P < 0.001, permutation test, Additional file 1: Figure S5a). This observation is possibly related to the broadest interquartile range of DNA curvature in coding sequences among these three genomic regions (Additional file 1: Figure S5b).

Mutations in human tumors are enriched in DNA regions with a smaller intrinsic DNA curvature. a Mutations are enriched in regions with a significantly smaller curvature in all 26 cancer types. Each dot represents a cancer type. P values were calculated based on the permutation test. P values were arbitrarily assigned to 0.001 when P < 0.001. b, c Examples in lung (b) and in kidney (c) showing that the average intrinsic DNA curvature of SNV-containing regions (red arrows) was significantly smaller than the random expectation (histogram showing 1000 permutations)
The large number of somatic mutations in tumor cells permitted a more robust test of the effect for nucleotide context. We found that DNA curvature negatively correlates with mutation rate when controlling for the trinucleotide (Additional file 1: Figure S6) or heptanucleotide context (Additional file 1: Figure S7). In addition, we performed logistic regression to predict whether a site has a somatic mutation in at least one cancer sample. Variables used in the regression model include the nucleotide at a site, six flanking nucleotides (three upstream and three downstream) around the site [16], and intrinsic DNA curvature of the 101-bp region from 50 bp upstream to 50 bp downstream of the site. Consistently, we found that DNA curvature was a negative predictor of somatic mutations, and the effect of it is comparable to a nucleotide substitution in the six flanking sites (Additional file 1: Figure S8). The type of nucleotide at the site was a strong predictor of somatic mutations in human tumor cells (Additional file 1: Figure S8), likely because variation in DNA methylation among CpG sites plays an important role in determining mutation rate [15]. In contrast, DNA methylation is virtually none in the budding yeast [33].
To determine if our results from somatic mutations in human tumors are applicable to germline mutations, we further retrieved 101,377 de novo point mutations identified from 1548 trios from Iceland [34]. Again, we observed a smaller DNA curvature around these mutations (P < 0.001, permutation test, Additional file 1: Figure S9). Taken together, DNA curvature is a robust predictor of non-uniform mutation rates in both yeast and humans.
Genetic manipulation of DNA curvature affects mutation rate
To further examine the causal effect of intrinsic DNA curvature on mutation rate, we designed four synonymous variants of URA3 (Additional file 1: Table S4), two with increased curvature and two with decreased curvature (Fig. 4a). We kept features that may influence local mutation rate such as GC content, codon usage, and predicted local mRNA structure largely unchanged (Additional file 1: Table S5) [13, 17, 26]. The expression levels of URA3 in these variants are also identical (Additional file 1: Figure S10).

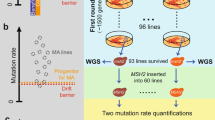
Changing the intrinsic DNA curvature in URA3 leads to altered mutation rate. a The distribution of the average intrinsic DNA curvature of genes in the yeast genome. The intrinsic DNA curvatures of five synonymous variants of URA3 are indicated by arrows. S1 and S2 (G1 and G2) are variants with a smaller (greater) intrinsic DNA curvature. b The schematic description of the experimental procedure for measuring the relative mutation rate of URA3 variants. c Reduction of intrinsic DNA curvature leads to an increase in the mutation rate of URA3. Outliers are not shown. P values were calculated from the one-tailed Mann-Whitney U test. d Similar to c, in a mismatch repair-deficient msh2 strain
We used an electrophoretic mobility shift assay to confirm that the intrinsic DNA curvature was altered in these variants [35,36,37]. Variants with a greater predicted intrinsic DNA curvature [19, 24] migrated more slowly than those with a smaller curvature (Additional file 1: Figure S11), presumably due to the different friction force that they encountered in the process of migration.
To determine if genetic manipulation of curvature alters mutation rate, we cultured cells with each of the five URA3 variants in SC media to allow mutations to accumulate, spread cells onto 5-FOA plates, and counted the number of colonies on each plate (Fig. 4b). We calculated the mutation rate of each variant from the fraction of plates without mutants [38] and found that variants with a 10% smaller intrinsic DNA curvature had a 70% higher mutation rate (Fig. 4c). It suggests that experimental decreasing DNA curvature increases mutation rate.
Intrinsic DNA curvature alters the mutation rate, not mismatch repair efficacy
There are two non-mutually exclusive mechanisms by which intrinsic DNA curvature can modulate the net mutation rate [9]. First, intrinsic DNA curvature may reduce the supply of mutations. Second, intrinsically curved DNA may facilitate the recruitment of mismatch repair-related proteins, which can increase the DNA repair efficacy [3, 9]. To determine if intrinsic DNA curvature reduces the supply of mutations or affects repair efficiency, we knocked out MSH2 and repeated the mutation accumulation experiment (Fig. 4b). In the absence of Msh2, the effect of DNA curvature on mutation rate is even larger; a 10% decrease in curvature results in a 100% increase in mutation rate (Fig. 4d). This observation suggests that the altered net mutation rate by DNA curvature is due to differences in the supply of mutations and not to differences in DNA repair efficacy.
DNA curvature is negatively correlated with mutagen sensitivity in human cancer cells
DNA curvature may reduce the mutation rate by making the DNA sequence less accessible to potential mutagens [27] or by affecting the fidelity of DNA polymerase itself, though this is unlikely, as DNA polymerase acts on single-stranded DNA. To distinguish these two mechanisms, we divided the SNVs in cancer cells into six categories based on mutation types and asked if the rate of mutation types that are sensitive to mutagens is more affected by DNA curvature. C>T transitions mainly result from the hydrolytic deamination on methylated cytosine [15, 39]. The rate of C>T transition reduced by 40% in DNA regions with a greater curvature (Fig. 5a). In contrast, this reduction in mutation rate was not observed for other mutation types (Fig. 5a). Furthermore, C>A transversions in lung cancer cells are mainly caused by polycyclic aromatic hydrocarbons in tobacco smoke [40,41,42]. C>A mutations are more affected by DNA curvature in lung cancer than they are in other types of cancer (Fig. 5b). Both biased distributions of C>T and C>A mutations suggest that curvature protects DNA from mutagens. Given the well-established role of DNA curvature in regulating protein-DNA interactions [20, 21], it is possible that DNA curvature promotes protein binding that makes DNA less accessible to mutagens [27].

DNA curvature suppresses mutations that are induced by mutagens. a The mutation rate of each of the six mutation types in cancer cells. Mutation rate was defined as the number of SNVs per cancer sample per nucleotide. x axis shows ten equally sized bins of DNA regions in the human genome sorted by intrinsic DNA curvature. b Mutation rates in lung cancer (including lung adenocarcinoma and lung squamous cell carcinoma)
Implications in evolutionary genomics
Understanding the variation in mutation rate is central to numerous questions in evolutionary genetics. Particularly, modeling the variability in mutation rate among sites of a genome is of key importance in studies of molecular evolution because it provides a null model that can be rejected when natural selection occurs. Sequence-intrinsic cis elements are more computationally tractable than trans factors in modeling mutation rate in molecular evolution studies, because with cis elements the expected mutation rate can be predicted directly from the surrounding sequences of a site [16]. For example, the evolutionary rates of genes have been extensively studied, and particularly, comparisons between those of essential and nonessential genes have been made [43,44,45,46,47]. Previous studies focused on the difference in the strength of negative selection and neglected the potential difference in mutation rate, presumably because the latter was hard to model. In this study, we discovered that a key DNA shape feature, intrinsic DNA curvature, modulated local mutation rate. Interestingly, we observed that essential genes exhibit a greater DNA curvature in both yeast (Additional file 1: Figure S12) and humans (Additional file 1: Figure S13), suggesting that they have a lower mutation rate. This observation urges the need of considering the difference in mutation rate when comparing evolutionary rate among genes.
Furthermore, the high-density fitness landscapes of random mutations on a gene have been extensively characterized in previous studies [48, 49], aiming to understand the trajectory of biological evolution. However, evolutionary trajectories are determined by natural selection acting on mutations. Inherent biases in the generation of the random mutations must therefore be taken into account. Our study on mutational landscape complements these previous studies on fitness landscapes and will significantly contribute to the ultimate understanding of evolutionary trajectories [50].
Conclusions
We found that the shape of the DNA double helix plays a major role in determining the local mutation rate. In particular, we identified a key feature, intrinsic DNA curvature, that determines the local mutation rate in both yeast and humans. We genetically manipulated the intrinsic DNA curvature and observed an altered mutation rate consistent with the genome-wide data. We showed that this effect is due to increased mutation rate, likely due to increased exposure to mutagens, and not due to differential efficacy of repair machinery. Taken together, our study extensively expands our knowledge of elements that regulate mutation rate by integrating the valuable information of DNA shape, and develops a new framework to understand evolution and tumorigenesis at a nucleotide resolution.
Methods
Characterization of the mutational landscape of URA3
A haploid S. cerevisiae strain derived from the W303 background, GIL104 (MATa URA3, leu2, trp1, CAN1, ade2, his3, bar1Δ::ADE2), was used to characterize the mutational landscape of URA3. Cells from a single colony were cultured in 5 ml SC media with uracil dropped-out (SC-uracil) at 30 °C for 24 h. Cells were then transferred into 5 ml fresh SC media (at an initial OD660 ~ 0.1) and grown for 24 h to accumulate mutations. ~ 5.0 × 107 cells were spread onto SC-uracil plates containing 1 g/l 5-FOA to select for loss-of-function mutants of URA3. A total of ~ 1000 ura3 variants were isolated from 5-FOA plates and were Sanger sequenced separately. PCR and Sanger sequencing primers are listed in Additional file 1: Table S6.
Calculation of the mutation rate and the values of DNA properties in URA3 and CAN1
We identified a total of 452 mutations in URA3 (Additional file 1: Table S1), including 5 synonymous mutations, 293 missense mutations, and 154 nonsense mutations. We focused on these 154 nonsense mutations in this study for the sake of accuracy in estimating mutation rate. To be specific, we need to count the number of potential loss-of-function mutation sites, which would be used to normalize the number of observed mutations and hence to calculate the mutation rate. The number of potential loss-of-function missense mutations was difficult to estimate because it remains elusive which missense mutations lead to a loss of function and which do not. Mutation rate was determined using overlapping windows with size equal to L nucleotides (L = 10, 20 …, or 100 bp, Additional file 1: Figure S3). The window slid for 10 nucleotides each movement. The value of a DNA shape feature was calculated based on the frequencies of all 16 possible combinations of dinucleotide in a region, following previous studies [19, 24].
Estimation of nucleosome occupancy
The wild-type S. cerevisiae strain (BY4741 URA3) was grown to log-phase in YPD (1% yeast extract, 2% peptone, and 2% dextrose) liquid medium. We performed nucleus isolation, micrococcal nuclease (MNase) digestion, and chromatin preparation as described previously [51], with the following modifications. We adjusted NP-S buffer to 0.5 mM spermidine, 0.075% (v/v) NP-40, 50 mM NaCl, 50 mM Tris-HCl pH 7.5, 5 mM MgCl2, and 5 mM CaCl2, and used 100 units of MNase to digest the nuclei for 5 min. We performed Protease K digestion and exacted the core particle DNA. Paired-end libraries were constructed using Illumina-compatible DNA-Seq NGS library preparation kit from Gnomegen and were sequenced with Illumina HiSeq 2500 (PE125, paired-end 2 × 125 bp). ~ 10.6 million clean reads were aligned to the S. cerevisiae genome using bowtie2 with default parameters [52]. Nucleosome occupancy of a nucleotide was defined as the number of read pairs uniquely mapped to the genome region covering the nucleotide. The raw sequencing data of MNase-seq have been deposited to the Genome Sequence Archive [53] in BIG Data Center (http://bigd.big.ac.cn/gsa), Beijing Institute of Genomics, Chinese Academy of Sciences, under accession number CRA000570.
Generation and analyses of the URA3 variants
We designed four synonymous variants of URA3 with different intrinsic DNA curvature (Additional file 1: Tables S4–S5). We estimated the minimum free energy (MFE) for all 20 nucleotide windows in the coding sequence with RNAfold [54] and defined the average MFE of them as the strength of the RNA secondary structure of a variant. Codon adaptation index (CAI) was calculated following our previous study [55]. Four URA3 variants were synthesized by Wuxi Qinglan Biotech, and the wild-type URA3 DNA sequence was amplified from S288C. Primers are listed in Additional file 1: Table S6. Each of the five variants was introduced into the chromosomal location of URA3 in BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) with homologous recombination.
We used electrophoretic mobility shift assay to confirm the difference in intrinsic DNA curvature of the five synonymous variants. We loaded an equal amount of PCR products of five variants into a 12% native polyacrylamide gel. We performed the electrophoresis experiment in the TBE buffer (89 mM Tris, 89 mM boric acid, and 2.5 mM EDTA, pH 8.0) for 12 h at 120 V.
Total RNA was extracted with hot acidic phenol (pH < 5.0) and was reverse transcribed with the GoScript™ reverse transcriptase. Quantitative PCR (qPCR) was carried out on the Mx3000P qPCR System (Agilent Technologies) using Maxima SYBR Green/ROX qPCR Master Mix. ACT1 was used as the internal control. Primers used are listed in Additional file 1: Table S6.
The variance-to-mean ratio of the numbers of colonies on the plates was much greater than 1 for each variant (Additional file 1: Figure S14), indicating that the number of colonies does not follow a Poisson distribution [38]. This suggests that the observed mutations most likely occurred in the liquid culture instead of on the plates. We used the non-parametric Mann-Whitney U test to compare the number of colonies among these strains. We also estimated the relative mutation rates in these variants from p0, the proportion of cultures with no mutants, in the wild-type background with the following equation [38].
Estimation of mutation rate in yeast mutation accumulation (MA) lines
A previous study identified ~ 1000 single nucleotide mutations by sequencing the genomes of five MA lines of a mismatch repair-deficient S. cerevisiae strain (BY4741 msh2::kanMX4) [30]. The mutation data from this study was used because the efficacy of purifying selection in MA experiments [17, 23] was further reduced in mutators. We analyzed the mutations supported by ≥ 20× coverage and retrieved 882 single nucleotide mutations that were identified in at least one of the five replicates from this study. As a control, we chose 882 random sites in the rest of the yeast genome and defined them as the pseudo-mutation sites. We calculated the average intrinsic DNA curvature around these pseudo-mutation sites and repeated this procedure for 1000 times. P values were calculated as the fraction of pseudo-mutation sets exhibiting a smaller average intrinsic DNA curvature than that of the observed mutation sites among 1000 permutations.
Estimation of mutation rate in humans
When multiple projects for a cancer type exist, we combined all SNVs in these projects. On average, ~ 100,000 SNVs were identified in a cancer type. For each cancer type, we calculated the average intrinsic DNA curvature of the flanking DNA sequences of all SNVs (from 50 bp upstream to 50 bp downstream of each SNV). We also randomly chose the same number of sites in the human genome and calculated the average intrinsic DNA curvature of their flanking sequences similarly. This procedure was repeated 1000 times to obtain the distribution of the expected average intrinsic DNA curvature. P values were calculated as the fraction of sets of random sites exhibiting a smaller average intrinsic DNA curvature than that of the observed SNV sites, among 1000 permutations. In TCGA, different methods were used to identify mutations (Mutect, Muse, Somaticsniper, and Varscan, Additional file 1: Figures S4, S6–S7). The SNVs in 5′ UTR, coding sequences, and 3′ UTR were also separately analyzed, with the expectation obtained by only sampling DNA sequences in the corresponding type of genomic regions. Because the number of SNVs in 5′ UTR and 3′ UTR were relatively small, SNVs in all cancer types were combined. In addition, 101,377 de novo point mutations in the human germline were retrieved from a previous study [34]. Permutation test were performed as described in cancer cells (Fig. 3).
References
Hodgkinson A, Eyre-Walker A. Variation in the mutation rate across mammalian genomes. Nat Rev Genet. 2011;12:756–66.
Schuster-Bockler B, Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012;488:504–7.
Frigola J, Sabarinathan R, Mularoni L, Muinos F, Gonzalez-Perez A, Lopez-Bigas N. Reduced mutation rate in exons due to differential mismatch repair. Nat Genet. 2017;49:1684–92.
Prendergast JG, Campbell H, Gilbert N, Dunlop MG, Bickmore WA, Semple CA. Chromatin structure and evolution in the human genome. BMC Evol Biol. 2007;7:72.
Lang GI, Murray AW. Mutation rates across budding yeast chromosome VI are correlated with replication timing. Genome Biol Evol. 2011;3:799–811.
Stamatoyannopoulos JA, Adzhubei I, Thurman RE, Kryukov GV, Mirkin SM, Sunyaev SR. Human mutation rate associated with DNA replication timing. Nat Genet. 2009;41:393–5.
Chen CL, Rappailles A, Duquenne L, Huvet M, Guilbaud G, Farinelli L, Audit B, d'Aubenton-Carafa Y, Arneodo A, Hyrien O, Thermes C. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res. 2010;20:447–57.
Weber CC, Pink CJ, Hurst LD. Late-replicating domains have higher divergence and diversity in Drosophila melanogaster. Mol Biol Evol. 2012;29:873–82.
Supek F, Lehner B. Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature. 2015;521:81–4.
Herman RK, Dworkin NB. Effect of gene induction on the rate of mutagenesis by ICR-191 in Escherichia coli. J Bacteriol. 1971;106:543–50.
Park C, Qian W, Zhang J. Genomic evidence for elevated mutation rates in highly expressed genes. EMBO Rep. 2012;13:1123–9.
Savic DJ, Kanazir DT. The effect of a histidine operator-constitutive mutation on UV-induced mutability within the histidine operon of Salmonella typhimurium. Mol Gen Genet. 1972;118:45–50.
Chen X, Yang JR, Zhang J. Nascent RNA folding mitigates transcription-associated mutagenesis. Genome Res. 2016;26:50–9.
Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–70.
Coulondre C, Miller JH, Farabaugh PJ, Gilbert W. Molecular basis of base substitution hotspots in Escherichia coli. Nature. 1978;274:775–80.
Aggarwala V, Voight BF. An expanded sequence context model broadly explains variability in polymorphism levels across the human genome. Nat Genet. 2016;48:349–55.
Zhu YO, Siegal ML, Hall DW, Petrov DA. Precise estimates of mutation rate and spectrum in yeast. Proc Natl Acad Sci U S A. 2014;111:E2310–8.
Blake RD, Hess ST, Nicholson-Tuell J. The influence of nearest neighbors on the rate and pattern of spontaneous point mutations. J Mol Evol. 1992;34:189–200.
Olson WK, Gorin AA, Lu XJ, Hock LM, Zhurkin VB. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc Natl Acad Sci U S A. 1998;95:11163–8.
Harteis S, Schneider S. Making the bend: DNA tertiary structure and protein-DNA interactions. Int J Mol Sci. 2014;15:12335–63.
Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B. The role of DNA shape in protein-DNA recognition. Nature. 2009;461:1248–53.
Wang X, Zhou T, Wunderlich Z, Maurano MT, DePace AH, Nuzhdin SV, Rohs R. Analysis of genetic variation indicates DNA shape involvement in purifying selection. Mol Biol Evol. 2018;35:1958–67.
Lang GI, Murray AW. Estimating the per-base-pair mutation rate in the yeast Saccharomyces cerevisiae. Genetics. 2008;178:67–82 https://doi.org/10.1534/genetics.107.071506.
Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, Nislow C. A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet. 2007;39:1235–44.
Bolshoy A, McNamara P, Harrington RE, Trifonov EN. Curved DNA without A-A: experimental estimation of all 16 DNA wedge angles. Proc Natl Acad Sci U S A. 1991;88:2312–6.
Wolfe KH, Sharp PM, Li WH. Mutation rates differ among regions of the mammalian genome. Nature. 1989;337:283–5.
Chen X, Chen Z, Chen H, Su Z, Yang J, Lin F, Shi S, He X. Nucleosomes suppress spontaneous mutations base-specifically in eukaryotes. Science. 2012;335:1235–8.
Smith T, Ho G, Christodoulou J, Price EA, Onadim Z, Gauthier-Villars M, Dehainault C, Houdayer C, Parfait B, van Minkelen R, et al. Extensive variation in the mutation rate between and within human genes associated with Mendelian disease. Hum Mutat. 2016;37:488–94.
Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, Olivier M. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat. 2016;37:865–76.
Fares MA, Keane OM, Toft C, Carretero-Paulet L, Jones GW. The roles of whole-genome and small-scale duplications in the functional specialization of Saccharomyces cerevisiae genes. PLoS Genet. 2013;9:e1003176 https://doi.org/10.1371/journal.pgen.1003176.
Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordonez GR, Bignell GR, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–6.
Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–20.
Capuano F, Mulleder M, Kok R, Blom HJ, Ralser M. Cytosine DNA methylation is found in Drosophila melanogaster but absent in Saccharomyces cerevisiae, Schizosaccharomyces pombe, and other yeast species. Anal Chem. 2014;86:3697–702.
Jonsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, et al. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22 https://doi.org/10.1038/nature24018.
Hagerman PJ. Sequence-directed curvature of DNA. Nature. 1986;321:449–50.
Koo HS, Wu HM, Crothers DM. DNA bending at adenine. Thymine tracts. Nature. 1986;320:501–6.
Ulanovsky LE, Trifonov EN. Estimation of wedge components in curved DNA. Nature. 1987;326:720–2.
Luria SE, Delbruck M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics. 1943;28:491–511.
Maki H. Origins of spontaneous mutations: specificity and directionality of base-substitution, frameshift, and sequence-substitution mutagenesis. Annu Rev Genet. 2002;36:279–303.
Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3:246–59.
Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8.
Polak P, Karlic R, Koren A, Thurman R, Sandstrom R, Lawrence M, Reynolds A, Rynes E, Vlahovicek K, Stamatoyannopoulos JA, Sunyaev SR. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature. 2015;518:360–4.
Hurst LD, Smith NG. Do essential genes evolve slowly? Curr Biol. 1999;9:747–50.
Hirsh AE, Fraser HB. Protein dispensability and rate of evolution. Nature. 2001;411:1046–9.
Wang Z, Zhang J. Why is the correlation between gene importance and gene evolutionary rate so weak? PLoS Genet. 2009;5:e1000329.
Liao BY, Scott NM, Zhang J. Impacts of gene essentiality, expression pattern, and gene compactness on the evolutionary rate of mammalian proteins. Mol Biol Evol. 2006;23:2072–80.
Zhang J, Yang JR. Determinants of the rate of protein sequence evolution. Nat Rev Genet. 2015;16:409–20.
Li C, Qian W, Maclean CJ, Zhang J. The fitness landscape of a tRNA gene. Science. 2016;352:837–40.
Puchta O, Cseke B, Czaja H, Tollervey D, Sanguinetti G, Kudla G. Network of epistatic interactions within a yeast snoRNA. Science. 2016;352:840–4.
He X, Liu L. EVOLUTION. Toward a prospective molecular evolution. Science. 2016;352:769–70.
Wal M, Pugh BF. Genome-wide mapping of nucleosome positions in yeast using high-resolution MNase ChIP-Seq. Methods Enzymol. 2012;513:233–50.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Wang Y, Song F, Zhu J, Zhang S, Yang Y, Chen T, Tang B, Dong L, Ding N, Zhang Q, et al. GSA: genome sequence archive. Genomics Proteomics Bioinformatics. 2017;15:14–8.
Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011;6:26.
Chen S, Li K, Cao W, Wang J, Zhao T, Huan Q, Yang YF, Wu S, Qian W. Codon-resolution analysis reveals a direct and context-dependent impact of individual synonymous mutations on mRNA level. Mol Biol Evol. 2017;34:2944–58.
Christie KR, Weng S, Balakrishnan R, Costanzo MC, Dolinski K, Dwight SS, Engel SR, Feierbach B, Fisk DG, Hirschman JE, et al. Saccharomyces genome database (SGD) provides tools to identify and analyze sequences from Saccharomyces cerevisiae and related sequences from other organisms. Nucleic Acids Res. 2004;32:D311–4.
Deutschbauer AM, Jaramillo DF, Proctor M, Kumm J, Hillenmeyer ME, Davis RW, Nislow C, Giaever G. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics. 2005;169:1915–25 https://doi.org/10.1534/genetics.104.036871.
Qian W, Ma D, Xiao C, Wang Z, Zhang J. The genomic landscape and evolutionary resolution of antagonistic pleiotropy in yeast. Cell Rep. 2012;2:1399–410 https://doi.org/10.1016/j.celrep.2012.09.017.
Qian W, Zhang J. Genomic evidence for adaptation by gene duplication. Genome Res. 2014;24:1356–62 https://doi.org/10.1101/gr.172098.114.
Chatr-Aryamontri A, Oughtred R, Boucher L, Rust J, Chang C, Kolas NK, O'Donnell L, Oster S, Theesfeld C, Sellam A, et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017;45:D369–79.
Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell. 2015;163:1515–26 https://doi.org/10.1016/j.cell.2015.11.015.
Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, Sabatini DM. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–101 https://doi.org/10.1126/science.aac7041.
Huang N, Lee I, Marcotte EM, Hurles ME. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154 https://doi.org/10.1371/journal.pgen.1001154.
Friedel M, Nikolajewa S, Suhnel J, Wilhelm T. DiProDB: a database for dinucleotide properties. Nucleic Acids Res. 2009;37:D37–40.
Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, Ledbetter DH, Maglott DR, Martin CL, Nussbaum RL, et al. ClinGen--the clinical genome resource. N Engl J Med. 2015;372:2235–42.
Duan C, Huan Q, Chen X, Wu S, Carey LB, He X, Qian W. Reduced intrinsic DNA curvature leads to increased mutation rate. Genome Sequence Archive. 2018. http://bigd.big.ac.cn/gsa/browse/CRA000570. The release date: 19 Apr 2018.
Acknowledgements
We thank Yuliang Zhang for technical support in data analysis, and Mengyi Sun and Jian-Rong Yang for critical reading of the manuscript.
Review history
The review history for this manuscript is available as Additional file 2.
Funding
This work was supported by grants from the National Natural Science Foundation of China to X.H. and W.Q. (91731302).
Availability of data and materials
Protein-protein interaction (PPI) data in yeast were downloaded from Saccharomyces Genome Database (https://www.yeastgenome.org/) [56]. Lists of essential genes and haploinsufficient genes were retrieved from a previous study [57]. Genes leading to significant growth reduction upon deletion were identified in a previous study with Bar-seq [58]. Duplicate genes in the yeast genome were defined in a previous study [59]. PPI data in humans were downloaded from Biogrid (https://thebiogrid.org/) [60]. Human essential genes were retrieved from two previous studies [61, 62], respectively. The list of haploinsufficient genes in humans were retrieved from a previous study [63]. The value of each dinucleotide for each DNA shape feature was obtained from the Dinucleotide Property Database (http://diprodb.leibniz-fli.de/ShowTable.php) [64]. The data of SNVs in cancer cells were retrieved from The Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov/) [32]. Chromosomal sequences surrounding these SNVs were retrieved from Ensembl release 87 (www.ensembl.org). Mutations in MECP2, NF1, and RB1 were retrieved from ClinGen (https://clinicalgenome.org/) [65] and mutations in TP53 were retrieved from IARC TP53 Database (http://p53.iarc.fr/) [29].
The raw sequencing data of this study have been deposited to the Genome Sequence Archive in BIG Data Center (http://bigd.big.ac.cn/gsa) under accession number CRA000570 [66].
Author information
Authors and Affiliations
Contributions
CD, LBC, XH, and WQ designed the experiments. CD, QH, and XC performed the experiments. CD, QH, and WQ analyzed the data. CD, SW, LBC, and WQ wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Test of the Poisson distribution of nonsense mutations in URA3. Figure S2. The effect of nucleotide at the potential nonsense site on mutation rate. Figure S3. The estimation of the average mutation rate and the values of DNA properties. Figure S4. The results in cancer cells were not affected by SNV calling methods. Figure S5. DNA curvature is negatively associated with mutation rate in coding sequences in human tumors. Figure S6. The results in cancer cells held after controlling for the trinucleotide context. Figure S7. The results in cancer cells held after controlling for the heptanucleotide context. Figure S8. Logistic regression for predicting the presence of a SNV in human tumors. Figure S9. De novo point mutations in the human germline are enriched in DNA regions with a smaller DNA curvature. Figure S10. Expression levels were not significantly different among URA3 variants. Figure S11. Electrophoretic mobility shift assay showing differences in DNA curvature among URA3 variants. Figure S12. Comparison of intrinsic DNA curvature among yeast genes. Figure S13. Comparison of intrinsic DNA curvature among human genes. Figure S14. The distribution of the number of colonies on 60 5-FOA plates for each of the five URA3 variants. Table S1. Numbers of plates containing a mutation in URA3. Table S2. Models on predicting the mutation rate of a potential nonsense site in CAN1. Table S3. Modeling the mutation rate of a potential nonsense site in four human genes. Table S4. DNA sequences of URA3 variants. Table S5. Features of five URA3 variants. Table S6. Primers used in this study. (DOCX 1081 kb)
Additional file 2:
Review history. (DOCX 41 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Duan, C., Huan, Q., Chen, X. et al. Reduced intrinsic DNA curvature leads to increased mutation rate. Genome Biol 19, 132 (2018). https://doi.org/10.1186/s13059-018-1525-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13059-018-1525-y