
Abstract
Through RNA-Seq analyses, we identified 137 genes that are missing in chicken, including the long-sought-after nephrin and tumor necrosis factor genes. These genes tended to cluster in GC-rich regions that have poor coverage in genome sequence databases. Hence, the occurrence of syntenic groups of vertebrate genes that have not been observed in Aves does not prove the evolutionary loss of such genes.
Please see related Research article: http://dx.doi.org/10.1186/s13059-014-0565-1 and Please see response from Lovell et al: https://www.dx.doi.org/10.1186/s13059-017-1234-y
Similar content being viewed by others


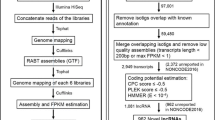
A recent paper reported that 274 protein-encoding genes were missing from sequencing data from 60 bird species [1]. Most of them were organized in conserved syntenic clusters in non-avian vertebrates, suggesting that their loss in the avian lineage had occurred through genomic deletions of gene blocks. This hypothesis was supported by another study reporting that 640 protein-encoding genes were missing from 48 bird genomes [2]; the authors of this second study made a similar suggestion that large segmentally deleted regions had been lost during microchromosome evolution in birds. However, our recent discovery of leptin genes with ~70% GC content in chicken and duck [3], and the new identification of 89 GC-rich genes [4], suggested an alternative hypothesis of a technical barrier to explain the ‘missing genes’. To further explore this, RNA-Seq data from visceral fat, hypothalamus, and pituitary tissues from two types of chickens, broilers and layers (Additional file 1: Table S1), were used for de novo transcriptome assembly and identification of novel genes.
The initial set of 588,683 transcripts obtained using Trinity [5] was reduced to 257,700 after removing transcripts that were expressed at low levels. We mapped the transcripts to the chicken reference genome build consistent with the previous studies [1, 2] using Blat and Blast, and retained 8395 sequences without alignments. These transcripts were then characterized on the basis of sequence similarity to known genes in other vertebrates using the Trinotate pipeline (https://trinotate.github.io), which searches for sequences encoding known protein domains, transmembrane domains, and signal peptides (Additional file 1: Tables S2 and S3a). Genes that were already known in chicken were removed by comparing their gene symbols with those in Ensembl (release 80), RefSeq, and Entrez Gene, resulting in 1878 novel gene-candidate transcripts representing 1063 genes (Additional file 1: Tables S3b and S4).
To increase specificity and to remove multiple transcript isoforms, we tested each transcript by reciprocal Blastn against the full transcriptome assembly (588,683 transcripts), and Blastx against the set of coding sequences predicted by TransDecoder (https://transdecoder.github.io), consisting of 111,457 sequences. The remaining set yielded 194 transcripts encompassing 190 distinct high-confidence genes (Additional file 1: Table S5). Through Blastn, we found that 55 loci had already been recovered as annotated genes in an updated genome build (Galgal5) released after the previous studies. In addition, 47 genes mapped to the genome but lacked annotations, while another 51 genes were annotated as uncharacterized or putative proteins (Additional file 1: Table S6). One discrepancy in annotation between our genes and Galgal5 was observed for the RSAD1 transcript, which was annotated as MYCBPAP in Galgal5. Closer inspection revealed that these two genes, which are close neighbors in the human genome, have been mistakenly merged into MYCBPAP in Galgal5. Therefore, we considered RSAD1 as a novel annotation (Additional file 1: Table S6).
Among the remaining 38 genes (Additional file 1: Table S6) with no sequence similarity to any genome build are the tumor necrosis factor (TNF) and nephrin (NPHS1), which have been reported as missing from birds in several studies (Table 1) but which are critically important in vertebrate biology and have extensively been studied in non-avian vertebrates (there are more than 130,000 publications in PubMed on TNF and 1300 on NPHS1). These genes were subjected to full-cDNA-sequence determination, exon characterization, RT-PCR validation, and expression profiling using RNA-Seq data from red junglefowl (Additional file 2: Figures S1 and S2; Additional file 2: Tables S9 to S12). The similarity in sequences, exon–intron junctions, and characteristic expression profiles confirmed the identification of chicken NPHS1 and TNF, thus resolving the long discussion as to why these genes have been missing from the genome assembly despite their established essential biological function in other species (for examples, see [6,7,8,9,10,11,12]).
Mass spectrometry analysis of fat tissue from the same chickens confirmed the identification of MEPCE, NPC1L1, PHF1, MRPS18, and SF3B2 at P < 0.01, and the expression of AMIGO1, CYAB, FKBP11, MGAT1, MOGS, MRI1, MTX1, POLR3D, PEA15, and TXNIP at P < 0.05 (Additional file 1: Tables S4, S5, and S8). To further validate the novel genes in the context of species phylogeny, we selected 11 genes with complete coding sequences predicted by TransDecoder (Additional file 3: Table S13) and at least four reported orthologous protein sequences in the NCBI protein database, for analysis of protein identity with the predicted chicken amino acid sequence using pBlast. As expected, the relative degrees of sequence identity were inversely correlated with evolutionary distance for most transcripts (r = –1 to –0.7), with three exceptions resulting from high conservation.
Comparing these genes to the genes previously reported as missing [1, 2, 6] recovered 74 overlapping gene symbols (Table 1). A higher proportion of the genes reported missing only in chickens was identified compared to those reported missing in all avian species (15% and 3–4.5%, respectively). The recovered transcripts had very high GC content (68%; Additional file 3: Figure S3b), further supporting the hypothesis that many of the genes that are currently missing from the draft genome eluded previous identification because of their high GC content [3, 4].
When exploring the location of novel genes recovered by the updated genome build, we observed that most genes (76%) were located on unplaced scaffolds, probably representing uncharacterized microchromosomes. Among those that mapped to known chromosomes, the majority (80%) were localized to microchromosomes, which are estimated to contain 50% of protein-coding genes in chickens [13]. Surprisingly, many of the mapped genes appeared in clusters. Mapping positions of the human orthologs demonstrated that the organization of 80% of the mapped novel genes was in syntenic clusters (Table 2). The strong tendency of these novel genes to cluster indicated their location in recalcitrant chromosomal regions with high GC content, primarily on microchromosomes. The methods used in this study are detailed in Additional file 4: Detailed materials and methods.
Conclusions
Our RNA-Seq study, combined with extensive bioinformatics analysis, recovered 191 novel genes that were missing from previous chicken assemblies, 38 of which are still not present in the most recent genome build (Galgal5), as well as an additional 47 that are at least partially present in Galgal5 but lacking proper annotation. The high GC content (68% on average), the microchromosomal location of the majority of the novel genes (80%) covered by Galgal5, and their high tendency to cluster into syntenic blocks (80%) suggest that the novel genes were not found in earlier analyses because of their position in GC-rich gene clusters, rather than due to chromosomal fragmentation and loss. In addition, the identification and characterization of NPHS1 and TNF, which are expected to be essential for avian physiology, and which are still missing from the latest genome build, emphasizes the importance of striving towards a repertoire of known and characterized genes that is as complete as possible.
Abbreviations
- NPHS1:
-
Nephrin
- TNF:
-
Tumor necrosis factor
References
Lovell PV, Wirthlin M, Wilhelm L, Minx P, Lazar NH, Carbone L, et al. Conserved syntenic clusters of protein coding genes are missing in birds. Genome Biol. 2014;15:565.
Zhang G, Li C, Li Q, Li B, Larkin DM, Lee C, et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science. 2014;346:1311–20.
Seroussi E, Cinnamon Y, Yosefi S, Genin O, Smith JG, Rafati N, et al. Identification of the long-sought leptin in chicken and duck: expression pattern of the highly GC-rich avian leptin Fits an autocrine/paracrine rather than endocrine function. Endocrinology. 2016;157:737–51.
Hron T, Pajer P, Paces J, Bartunek P, Elleder D. Hidden genes in birds. Genome Biol. 2015;16:164.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52.
Dakovic N, Terezol M, Pitel F, Maillard V, Elis S, Leroux S, et al. The loss of adipokine genes in the chicken genome and implications for insulin metabolism. Mol Biol Evol. 2014;31:2637–46.
Miner JH. Life without nephrin: it’s for the birds. J Am Soc Nephrol. 2012;23:369–71.
Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65.
Wagner N, Morrison H, Pagnotta S, Michiels JF, Schwab Y, Tryggvason K, et al. The podocyte protein nephrin is required for cardiac vessel formation. Hum Mol Genet. 2011;20:2182–94.
Uysal B, Donmez O, Uysal F, Akaci O, Vuruskan BA, Berdeli A. Congenital nephrotic syndrome of NPHS1 associated with cardiac malformation. Pediatr Int. 2015;57:177–9.
Li X, Chuang PY, D’Agati VD, Dai Y, Yacoub R, Fu J, et al. Nephrin preserves podocyte viability and glomerular structure and function in adult kidneys. J Am Soc Nephrol. 2015;26:2361–77.
Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, et al. Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–82.
Smith J, Bruley CK, Paton IR, Dunn I, Jones CT, Windsor D, et al. Differences in gene density on chicken macrochromosomes and microchromosomes. Anim Genet. 2000;31:96–103.
Yaoita E, Nishimura H, Nameta M, Yoshida Y, Takimoto H, Fujinaka H, et al. Avian podocytes, which lack nephrin, use adherens junction proteins at intercellular junctions. J Histochem Cytochem. 2016;64:67–76.
Magor KE, Miranzo Navarro D, Barber MR, Petkau K, Fleming-Canepa X, Blyth GA, Blaine AH. Defense genes missing from the flight division. Dev Comp Immunol. 2013;41:377–88.
Acknowledgment
We thank Mr Mark Ruzal for growing the chickens. The study was supported by the ERC project BATESON (awarded to LA), Israel Academy of Sciences 876/14, and by the Chief Scientist of the Israeli Ministry of Agriculture 0469/14 (awarded to MFE and ES).
Availability of data and materials
The raw sequences that were used to build the trinity transcripts of the novel genes, as well as the cDNA sequences of the chicken NPHS1 and chicken and turkey TNF, are available in the ENA BioProject repository [PRJEB13623, www.ebi.ac.uk/ena/data/view/PRJEB13623].
Authors’ contributions
SB performed the transcript assembly and produced the novel gene lists. ES extended and characterized the NPHS1 and TNF predicted cDNAs and proteins. MFE and SY performed the biological experiments, prepared the RNA for sequencing, confirmed the deduced cDNA sequences of NPHS1 and TNF by RT-PCR, and performed the NPHS1 and TNF expression profiling. KP and SCB performed the MS analysis. MG helped to design the bioinformatic approaches. MFE, SB, and LA designed the experiments and wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
All animal procedures were carried out in accordance with the National Institutes of Health Guidelines on the Care and Use of Animals and Protocol IL536/14, which was approved by the Animal Experimentation Ethics Committee of the Agricultural Research Organization, Volcani Center, Rishon, Israel.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1:
Overview of the RNA-Seq data and filtration of the novel gene candidates. Table S1. Information about the RNA-Seq data. Table S2. The initial set of 2810 candidate novel transcripts. Table S3. Annotation, characterization, and filtering of the novel transcripts. Table S4. The intermediate set of 1878 transcripts representing 1063 candidate novel genes. Table S5. The high-confidence set of 194 transcripts representing 191 novel genes. Table S6. The 191 novel genes not included in Galgal4; 54 of these are correctly annotated while 137 are missing or lack correct annotation in Galgal5. Table S7. Characterization of the novel genes according to predicted cellular localization. Table S8. Identification of the novel genes in Galgal5 genome assembly and by Mass-Spec analysis in adipose tissue. (XLS 2362 kb)
Additional file 2:
Characterization of NPHS1 and TNF. Figure S1. Predicted full length cDNA sequence of NPHS1 and its characterization. Figure S2. Predicted full length cDNA sequence of TNF and its characterization. Table S9. Coding sequence of chicken NPHS1 and TNF predicted transcripts. Table S10. List of NPHS1 and TNF exons in human, turtle, and chicken. Table S11. List of primers used for RT-PCR. Table S12. Probes used for expression profiling in the Sequence Read Archive (SRA) database. (PDF 1672 kb)
Additional file 3:
Characterization of the high confidence novel genes. Table S13. Phylogenetic analysis of representative novel genes. Figure S3. Characterization of the novel transcripts. (PDF 232 kb)
Additional file 4:
Detailed materials and methods. Animals and tissue sampling. RNA-seq. Bioinformatic analysis. RT-PCR. Mass spectrometry analysis (MS). (PDF 283 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bornelöv, S., Seroussi, E., Yosefi, S. et al. Correspondence on Lovell et al.: identification of chicken genes previously assumed to be evolutionarily lost. Genome Biol 18, 112 (2017). https://doi.org/10.1186/s13059-017-1231-1
Published:
DOI: https://doi.org/10.1186/s13059-017-1231-1