
Abstract
Mucopolysaccharidoses (MPS) are a group of lysosomal storage disorders caused by a deficiency in lysosomal enzymes catalyzing the stepwise degradation of glycosaminoglycans (GAGs). The current therapeutic strategies of enzyme replacement therapy and allogeneic hematopoietic stem cell transplantation have been reported to reduce patient morbidity and to improve their quality of life, but they are associated with persistence of residual disease burden, in particular at the neurocognitive and musculoskeletal levels. This indicates the need for more efficacious treatments capable of effective and rapid enzyme delivery to the affected organs, especially the brain and the skeleton. Gene therapy (GT) strategies aimed at correcting the genetic defect in patient cells could represent a significant improvement for the treatment of MPS when compared with conventional approaches. While in-vivo GT strategies foresee the administration of viral vector particles directly to patients with the aim of providing normal complementary DNA to the affected cells, ex-vivo GT approaches are based on the ex-vivo transduction of patient cells that are subsequently infused back. This review provides insights into the state-of-art accomplishments made with in vivo and ex vivo GT-based approaches in MPS and provide a vision for the future in the medical community.
Similar content being viewed by others
Background
Mucopolysaccharidoses (MPS) are a group of 11 lysosomal storage diseases (LSDs) caused by a deficiency of enzymes involved in lysosomal breakdown of the glycosaminoglycans (GAGs)—heparan sulfate (HS), dermatan sulfate (DS), keratan sulfate (KS), chondroitin sulfate (CS), and hyaluronic acid [1, 2]. Consequently, these disorders are associated with the progressive accumulation of GAGs within the blood, connective tissues, and multiple organs. Clinically, MPS are widely heterogeneous and are characterized by a multisystem involvement affecting the liver, spleen, kidney, bone, cartilage, eyes, and central nervous system (CNS) [1, 2]. These diseases are autosomal recessive, except for the X-linked MPS II, and most of them have a variable phenotype from mild to severe. Symptoms develop within the first years of life and, without treatment, patients with the most severe forms typically succumb to disease complications within the first two decades of life. For this reason, timely diagnosis and early therapeutic intervention are crucial to prevent both somatic and CNS pathology.
Current therapeutic approaches for these disorders are directed towards supplying functional enzymes with the goal of lowering GAGs in tissues. These strategies rely on the mechanism of cross-correction [3], which is based on the fact that soluble lysosomal enzymes can be taken up by mannose-6-phosphate receptor-mediated endocytosis into affected cells. At present, enzyme replacement therapy (ERT) and allogeneic hematopoietic stem cell transplantation (HSCT) are the standard treatments for the majority of MPS diseases [4, 5]. Currently, ERT is approved exclusively for MPS I, MPS II, MPS IVA, and MPS VI [6, 7], although the first human treatment with investigational ERT in an MPS VII patient was recently reported [8]. No approved treatment is currently available for the MPS III disorders. Overall, ERT has proven to be effective in controlling most of the visceral manifestations of the diseases, while bone disease, heart valve disease, and corneal opacity have a tendency to be resistant [9]. In particular, CNS involvement has a tendency to be resistant to ERT because of the inability of the lysosomal enzymes to cross the blood–brain barrier (BBB) which impedes the passages of large therapeutic molecules into the brain. Furthermore, ERT has limitations since recombinant enzymes used for ERT can cause immunogenic responses that may translate into lack of efficacy [4, 5]. HSCT was used for the first time for the treatment of an inborn error of metabolism more than 30 years ago in an MPS IH (Hurler syndrome) patient [10]. The principle consists of the possibility of providing the patient with a permanent source of the missing enzyme which is represented by the engrafted donor-derived hematopoietic stem cells (HSCs) and their progeny. As HSCT also enables engraftment of donor-derived microglial cells in the brain, where they can locally produce the enzyme, this treatment, differing from the intravenous administration of enzymes, has the potential to treat CNS manifestations. HSCT has since become the standard of care for MPS IH patients. The availability of international guidelines for allogeneic HSCT in MPS, including high-resolution molecular human leukocyte antigen (HLA) typing for the selection of umbilical cord blood (UCB) units, individualized conditioning regimens, and supportive care, have contributed to render the transplant much safer, with transplant-related mortality < 10% [11, 12]. Overall, allogeneic transplantation is associated with a significant improvement in the survival rate and visceral manifestations, but has also revealed important morbidity due to residual disease burden, in particular at the musculoskeletal and neurocognitive levels [11,12,13,14,15].
Therefore, current transplantation strategies are only partially successful in treating MPS, probably due to the limited efficacy of the protein provided through hematopoiesis which allows only normal enzyme levels to be reached [11, 12]. In addition, the time at which the transplant procedure is performed could be too late to prevent pre-existing or progressive organ damage [16] and, for this reason, the development of safer conditioning regimens that could be employed at earlier ages is a critical accomplishment [17]. Moreover, the advent of new-born screening procedures may prove a major step forward in early identification and treatment of individuals with MPS [18].
Based on these premises, over the last years novel experimental therapies for MPS, including gene therapy (GT), substrate reduction therapy (SRT), anti-inflammatory therapy, and pharmacological chaperone therapy [4, 5, 19], have been investigated. In particular, this review focuses on the scientific evidence demonstrating that GT provides an achievable therapeutic option for MPS disorders. The two main categories of somatic GT consist of 1) the in-vivo infusion of viral vector particles with the aim of transferring normal complementary DNA to the affected cells enabling them to express the missing protein, or 2) the ex-vivo transduction of patient cells that are subsequently infused back. The first form is called in-vivo GT because the vector particles are administered directly to patients. In the ex-vivo procedure, patient cells are cultured in the laboratory, exposed to the viral vector that is carrying the desired gene, and then returned to the patient.
In-vivo GT approaches
Two important aspects allow MPS to be considered a suitable target for in-vivo GT approaches: monogenic recessive disorders, and that a small amount of therapeutic enzymes is able to improve the somatic and CNS pathology [19]. In-vivo GT is based on the concept of providing a functional copy of the defective gene to slow or reverse the disease state. A variety of different viral vectors, such as adenovirus, adeno-associated virus (AAV), retrovirus (RV), and lentivirus (LV), have undergone preclinical and clinical testing for their ability to mediate phenotypic improvements following their systemic, CNS direct, and intra-cerebrospinal fluid (CSF) injections in MPS animal models [4, 19, 20].
Among viral vectors for gene replacement, AAV vectors are those most used for in-vivo gene transfer since they are safe, provide significantly long transgene expression, and may be generated with variable serotypes allowing efficient delivery of therapeutic genes to different target tissues [21, 22]. Therefore, here, we will review mainly in-vivo GT studies based on AAV-mediated gene delivery. Viral vectors may be delivered using different administration routes (Fig. 1). Direct administration to the CNS represents a suitable delivery strategy when the CNS is the major therapeutic target. Direct CNS delivery may be achieved by either intracerebral or intrathecal/intraventricular injection in which the therapeutics access the brain, respectively, via parenchyma or intra-CSF. Intra-CSF delivery may also target somatic organs due to leakage of viral vectors in the blood stream. Direct CNS-targeting approaches generally represent high-invasive approaches for human therapeutic application. Systemic injection via the intravascular route is a noninvasive delivery strategy that represents the elective way to reach peripheral organs and tissues. Nevertheless, since every neuron in the brain is perfused by its own blood vessel, the intravenous administration of the therapeutic molecule can virtually reach all brain regions [23]. However, the BBB is not permeable to all the molecules and might impede effective delivery of therapeutic agents [24]. Thus, in general, while the somatic organs are efficiently targeted by a therapeutic molecule upon intravascular administration, the CNS instead is little or not targeted. However, as described below, different strategies may be employed to make intravascular-based delivery routes capable of overcoming the BBB obstacle.

Delivery routes in in-vivo GT approaches for MPS. Approaches for in-vivo GT of MPS may employ two different routes of viral vector administration. In direct central nervous system (CNS) administration routes, the viral vectors access the CNS via parenchyma (intracerebral injections) or via cerebrospinal fluid (CSF) (intrathecal/intraventricular injections). CNS is primarily targeted by these approaches. However, leakage of vectors in the bloodstream may result in the targeting and correction of somatic organ pathology. CNS direct approaches are generally invasive for clinical purposes. The second group of approaches is instead based on systemic injections (often directly into the blood stream) of viral vectors. These approaches efficiently target somatic organs and tissues. However, different strategies may be used to make these approaches capable of overcoming the blood–brain barrier (BBB) obstacle and treat the brain. Systemic approaches are minimally invasive and, in principle, more suitable for clinical purposes
Intracerebral injection
AAV viral vector administration in a specific area of the brain parenchyma has been extensively used in small and large animal models of MPS I [25], MPS IIIA [26], MPS IIIB [27], and MPS VII [28], where this strategy was able to rescue primary and secondary storage, thus preventing functional deficits. The promising results obtained in the preclinical studies in an MPS IIIA mouse model led to a phase I/II clinical trial in MPS IIIA patients (Lysogene) [29]. Four children received intracerebral injections of an AAV vector serotype rh.10-SGSH-IRES-SUMF1 delivered bilaterally to the white matter anterior, medial, and posterior to the basal ganglia at two different depths in a stereotaxic device at a dose of 7.2 × 1011 viral genomes/patient. Before treatment, all children were able to walk, but their cognitive abilities were abnormal and had declined; three out of four patients showed brain atrophy. The procedure was well tolerated with an absence of adverse events related to the injected product. At 1-year follow-up, neuropsychological evaluations suggested a possible, although moderate, improvement in behavior, attention, and sleep in three patients, whereas brain atrophy was stable in two patients but progressed in the others (Table 1) [29]. The high invasiveness of the surgical procedure and the limited distribution of the lysosomal enzymes to the injection site represent the major drawbacks of this approach. A phase II/III clinical trial based on the multisite delivery of AAV rh.10 encoding only N-sulfoglucosamine sulfohydrolase (SGSH) has been planned by Lysogene.
Intra-CSF injection
The intra-CSF AAV vector administration was recently proven to be efficacious in correcting somatic and CNS pathology in different MPS disorders in preclinical models. The main advantage of this strategy is the possibility of injecting the vector in the ventricles (i.e., lateral ventricle administration) or subarachnoid spaces (i.e., cisterna magna and intrathecal lumbar injections) filled with the CSF, thus reaching all the CNS structures surrounded by the CSF. Moreover, the leakage into the bloodstream makes these approaches capable of also targeting somatic tissues. Different examples of intra-CSF injections report the widespread CNS transduction and rescue of phenotypic aspects in MPS I [30], MPS IIIA [31, 32], MPS IIIB [33], and MPS VII [34] small and large animal models using different AAV serotypes. Among the AAV serotypes tested, the serotype 9 has been shown to be the most promising [35]. This, together with the safety of the lateral ventricle surgical procedure has recently led to approval of a clinical trial based on AAV9-mediated injection into the lateral ventricles of MPS IIIA patients (ESTEVE and Universitat Autònoma de Barcelona (UAB)) (Table 1). Clinical trials based on similar approaches are planned for MPS IIIB, MPS I, and MPS II (ESTEVE, UAB, and REGENXBIO).
However, intra-CSF-based approaches have some disadvantages, such as limited long-term gene expression and toxicity, factors that have to be considered before translating therapeutic strategies based on intra-CSF injection into clinical protocols.
Systemic administration
The intravenous liver-targeting strategy represents a noninvasive GT approach for the treatment of MPS. AAV vector serotypes with high tropism to the liver and liver-specific promoters have been tested to target the liver and convert it into a “factory” organ for the production and secretion of supraphysiological amounts of the lysosomal enzymes. The secreted enzymes traveling through the blood circulation reach the peripheral affected organs and are trafficked to the lysosomes. These approaches were successfully employed for the treatment of somatic pathology in MPS I and MPS VI animal models [36, 37], and a phase I/II clinical trial is currently ongoing for MPS VI patients (The MeuSIX consortium). This trial is based on AAV vector serotype 8 with liver-specific thyroxine-binding globulin (TBG) promoter driving the expression of the human ARSB gene. Vectors will be administered into a peripheral vein at three different doses (low dose, 2 × 1011 gc/kg; intermediate dose, 6 × 1011 gc/kg; high dose, 2 × 1012 gc/kg). However, the presence of the BBB makes liver-targeting strategies less suitable for the treatment of neuropathology in MPS. Indeed, intravenous administration of a lysosomal enzyme is able to markedly decrease the abnormal storage in non-neurological tissue, but has no or little effect on CNS pathology in disease mouse models [38]. The development of systemic therapeutic strategies that can overcome the major obstacle posed by the BBB represents the real challenge to establishing efficacious and safe therapies for MPS involving the CNS [39]. An approach to cross the BBB is to modify the therapeutic enzymes so that they can access one of the physiological transport systems localized within the BBB responsible for the delivery of molecules from the blood to the brain. Over the last years, a number of strategies have been developed to engineer the lysosomal enzymes with BBB-targeting motifs. These modified versions of the enzymes have been used in preclinical studies in MPS IIIA, MPS IIIB, MPS VII, MPS I, and MPS II animal models [39, 40]. In MPS IIIA, for example, two different strategies have been used to engineer the sulfamidase (the enzyme defective in MPS IIIA). In one, the binding domain of the apolipoprotein B, a protein that is normally transported across the BBB by low-density lipoprotein receptor (LDLR) binding, was fused to the C-terminal of the sulfamidase to allow the enzyme to be transcytosed through the BBB using the LDLRs on the BBB as a port to entry the brain. AAV vector serotype 8 (this serotype displays high liver tropism) encoding the chimeric sulfamidase was used as a vehicle to selectively target the liver via intravascular injection and convert it into a factory organ to produce the chimeric enzyme. Moreover, to increase the secretion of the sulfamidase form in the liver, the enzyme was further modified at the N-terminal with an alternative signal peptide derived from the highly secreted protein iduronate-2-sulfatase. This approach was successful in reverting neuropathology in MPS IIIA mice [41]. In a second approach, a BBB-penetrating form of sulfamidase was produced by re-engineering the enzyme as an IgG fusion protein, where the IgG domain is a monoclonal antibody (mAb) against the human insulin receptor (HIR). The HIRmAb domain of the HIRmAb-SGSH fusion protein acts as a molecular “Trojan horse” to ferry the fused enzyme across the BBB [42].
New attractive systemic AAV-mediated gene therapeutic approaches for the treatment of neuropathology are based on the use of specific AAV serotypes (e.g., AAV9, AAVrh10, AAVrh8) that are able to cross the BBB and target the CNS [43, 44]. A clinical trial based on AAV9 delivered intravascularly is currently under evaluation for MPS IIIA and is planned for MPS IIIB (Abeona Therapeutics). Preliminary data are now available for the first MPS IIIA patients treated with AAV9-SGSH. These data show a dose- and time-dependent CNS HS reduction together with neurocognitive benefit in the higher bar (older, low-dose; cohort 1), which implies benefit could be superior in cohort 2 (younger, high-dose).
In summary, AAV-mediated in-vivo GT for MPS is rapidly advancing towards the development of clinical protocols. However, some aspects, such as dosage of the viral vectors and their potential toxicity, have to be thoroughly understood before routinely applying these strategies for clinical management of patients. Another important field of investigation in in-vivo GT of genetic diseases is the possibility of directly modify defective genes by site-specific in vivo genome editing. Genome editing mediated by zinc finger nucleases (ZFN), transcription activator-like effector, or CRISPR/Cas9 nucleases have been explored for a number of genetic diseases at both the preclinical and clinical level [45]. Therapeutic strategies based on targeted insertion of the functional gene at the albumin locus in hepatocytes through in-vivo ZFN-mediated genome editing are currently being explored for attenuated forms of MPS I and MPS II (Sangamo BioSciences) and it is likely that these strategies will broaden clinical application in the near future.
Ex-vivo GT approaches
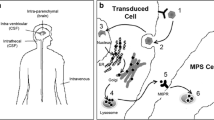
Ex-vivo GT entails the procurement and purification of target cells (i.e., CD34+ HSCs in the case of HSC-GT) that are transduced in vitro with a viral vector to express the therapeutic gene. Subsequently, the gene-corrected cells are returned to the patient after a chemotherapy-based preparative regimen of various intensity according to the disease treated [46], where they can engraft and restore the healthy phenotype [47,48,49,50] (Fig. 2). Both retroviral (RV) and lentiviral (LV) vectors that are able to integrate into the host genome can be employed to correct the altered gene in the HSCs and in their progeny. It has been demonstrated that the use of self-inactivating LVs is associated with a lower risk of insertional mutagenesis, and no events of leukemic transformation have been reported so far in patients treated in HSC-GT trials with this approach [51]. Moreover, LVs have the advantage of infecting nondividing cells and permitting higher levels of gene marking [52].

Ex-vivo GT approach for MPS. In ex-vivo GT approaches, patient cells are collected and stem cells are isolated; thereafter, they are mixed with the viral vector in which the therapeutic gene has been inserted. The final transduced stem cells are later re-infused in the patient after administration of a conditioning regimen and, by engrafting in the patient tissues, are able to restore the healthy phenotype. Modified from Penati et al., J Inherit Metab Dis. 2017, in press
Ex-vivo GT strategies have been tested in animal models and in clinical trials in LSDs, and in particular in MPS. RV-based HSC-GT proved to be effective in restoring enzyme activity and providing therapeutic benefit to visceral organs in MPS I mice; however, the CNS pathology was not sufficiently corrected, likely due to insufficient enzyme production by the engineered cells [53]. An LV-based approach has been employed to express alpha-l-iduronidase (IDUA) in an erythroid-specific manner by the use of a lineage-specific promoter in an MPS I mouse model. While gene-modified erythrocytes effectively released the enzyme into the circulation which was then distributed to the affected tissues, the neurological outcome was not improved compared with allogeneic HSCT [54]. The possibility of preventing and correcting neurological manifestations in mouse models of LSD by transplanting LV-transduced HSCs has also been reported for metachromatic leukodystrophy (MLD) and globoid cell leukodystrophy (GLD) [55,56,57]. Using this approach, microglia replacement by gene-corrected cells provided a population of efficient scavenger cells within the CNS with the potential to clear the accumulated substrate and which represents a new and effective source of bioavailable enzyme in the brain [55, 56]. In particular, in the mouse model of GLD, the infusion of HSCs corrected with an LV vector encoding for the galactocerebrosidase (GALC) gene was associated with reconstitution of the missing enzymatic activity in the liver and brain, together with improved survival and amelioration of the disease phenotype in the affected animals [57]. In the MLD mouse model, LV-based HSC-GT, but not transplantation of wild-type (WT) HSCs, was associated with prevention and correction of the neurological manifestations [55, 56]. In the MPS II mouse model, an LV-based HSC-GT approach was able to correct and prevent neuronal manifestations by ameliorating lysosomal storage and autophagic dysfunction in the brains of MPS II mice [58]. After transplantation of LV-transduced HSCs, increased enzyme activity was measured both in visceral organs and the CNS compared with naive mice, together with decreased levels of GAGs.
Based on these preclinical data, the therapeutic potential of LV-based HSC-GT has been tested in patients affected by MLD [49, 59] with the rationale to express supranormal levels of the therapeutic enzyme by gene-modified HSCs, particularly at the level of organs that are more difficult to reach, i.e., the CNS. The results of the first nine treated patients within a phase I/II study showed stable engraftment of the transduced HSCs in the bone marrow (BM) and peripheral blood of patients resulting in supranormal ARSA activity in hematopoietic lineages and in the CSF [49, 59]. The presence of ARSA activity in the CSF represents indirect evidence that HSC-derived cells have migrated to the brain and produced the enzyme locally. These findings were associated with a substantial therapeutic benefit of the procedure; better results were obtained in children who were treated when pre- or very early symptomatic. These data provide the first formal proof in humans that HSC-GT could represent a valuable therapeutic option for LSDs and constitutes an interesting platform for testing similar strategies in MPS disorders.
Indeed, published data on MPS IH patients treated with ERT and/or allogeneic HSCT indicate the need for a more efficacious treatment capable of early and higher levels of delivery to the affected organs, especially the brain and the skeleton. Indeed, allogeneic HSCT of MPS IH patients has been reported to significantly reduce patient morbidity and to improve their quality of life, but the therapeutic effect on bone and CNS lesions remains limited [15]. The musculoskeletal manifestations often require orthopedic surgical interventions after allo-HSCT and still deteriorate and impact the quality of life in most transplanted patients, probably due to the limited penetration of the expressed enzyme into the tissues [12, 14].
According to these premises, and taking advantage of the technology and expertise gained in the context of MLD, LV-driven supranormal IDUA reconstitution in HSCs and their progeny was tested at SR-TIGET with the aim of improving the outcome of the neurological and skeletal manifestations in MPS I mice. Therefore, MPS I mice were challenged with the transplantation of WT and LV-transduced Idua−/− HSCs in which enzymatic activity was restored to supranormal levels [60]. Results showed robust and effective delivery of the functional IDUA enzyme to diseased tissues, including the CNS where supranormal enzymatic activity was measured. This finding is particularly relevant in light of the inability of transplanted HSCs from normal WT donors (WT HSCT) to deliver comparable amounts of enzyme to the brain. Metabolic correction of the affected tissues was also demonstrated, as shown by the clearance of accumulated GAGs within hematopoietic and nonhematopoietic cells; this finding suggests the occurrence of active secretion of the functional enzyme by the gene-corrected progeny of the transplanted cells and its reuptake by the resident populations. A long-term toxicology/genotoxicity study was also conducted in MPS I mice under Good Laboratory Practice (GLP) conditions, employing an IDUA-LV produced under large-scale clinical grade conditions and by performing secondary transplantation into MPS I mice [61]. The study confirmed the long-term efficacy and safety of the approach in a rigorous testing environment. Indeed, stable engraftment of gene-modified cells was found, as well as sustained transgene expression resulting in the reconstitution of the defective IDUA activity at above WT levels in most of the GT-treated mice. Moreover, lack of evidence of any toxic or tumorigenic potential of IDUA-LV-transduced murine HSCs in primary and secondary transplant recipients was demonstrated. In a biodistribution study performed under GLP conditions, the repopulation of immunodeficient NSG mice by human UCB-derived CD34+ cells transduced with IDUA-LV was tested and compared with mock-transduced cells. Results confirmed stable IDUA overexpression in the human graft and in the myeloid progeny, as well as no functional impairment of human HSCs overexpressing IDUA, and the absence of recombinant replication-competent LV [61]. Importantly, BM CD34+ cells from MPS I patients behaved similar to those obtained from volunteer donors in terms of transducibility by IDUA-LV and reconstitution and overexpression of IDUA activity, as well as with regards to their clonogenic potential and capacity to repopulate immunodeficient mice [61].
Altogether, the significant therapeutic benefit observed in MLD patients and in MPS I mice and the data on the safety and tolerability of the approach both in mice and humans support the clinical testing of HSC-GT in MPS IH patients with the goal of augmenting the benefit of allogeneic HSCT, especially at the levels of the bone and CNS. Moreover, compared with allograft, the autologous procedure is associated with a reduced transplantation-related morbidity and mortality and, in particular, avoids the risks of allo-immune complications, i.e., rejection and graft-versus-host disease (GVHD). A phase I/II clinical trial is being implemented at SR-TIGET with the aim of testing the feasibility and safety of HSC-GT in MPS IH patients (Table 1). The trial will aim to achieve supranormal IDUA enzyme levels in patient blood cells to improve and/or prevent damage in visceral organs, CNS, and the skeleton. Concerning the skeletal tissue, the possibility that expression of supranormal enzyme levels might also favor the cross-correction of bone resident cells, such as mesenchymal stromal cells and osteoclasts, which have been reported to play a role in the skeletal manifestations of the disease, will be also investigated [62, 63].
MPS IIIA, an LSD caused by mutations in SGSH and resulting in HS accumulation and progressive neurodegeneration, is another potentially appealing target for which an LV-based, ex-vivo HSC-GT approach, similar to MPS IH, has been developed. Initial preclinical studies showed neurological improvement in the mouse model only when LV gene transfer was coupled to transplantation of WT cells. Subsequently, the use of an LV encoding for a codon-optimized therapeutic gene (SGSH) under the control of a myeloid-specific promoter to improve brain expression via monocyte/microglial specificity led to transgene overexpression and disease correction. In particular, the myeloid-specific promoter CD11b gave significantly higher monocyte/B-cell expression than PGK or CD18 promoters. When autologous MPS IIIA HSCs transduced with either LV-PGK-coSGSH or LV-CD11b-coSGSH vectors expressing codon-optimized SGSH were transplanted into MPS IIIA mice, the use of the latter vector was associated with significantly higher brain expression, normalization of MPS IIIA behavior, brain HS, and neuroinflammation [64]. The use of the myeloid-specific promoter CD11b to control the expression of the SGSH gene represents the main difference in this approach as compared with the strategy proposed for MPS IH patients.
A phase I/II clinical trial of HSC-GT for MPS IIIA, sponsored by Orchard Therapeutics Ltd., is planned for 2018 (Table 1). Enrolled patients will undergo a conditioning regimen based on myeloablative doses of busulfan, followed by infusion of autologous HSCs transduced with an LV vector expressing SGSH under the control of the CD11b myeloid-specific promoter. Safety and efficacy of the administration of the genetically modified HSCs will be evaluated.
In summary, over the last few years substantial progress has been made in HSC-GT for LSDs. Preclinical studies based on ex-vivo approaches for MLD, MPS IH, and GLD have shown that these strategies may have therapeutic impact on disease outcome. Moreover, a phase I/II LV-mediated HSC-GT protocol for MLD has suggested clinical benefit in late infantile presymptomatic patients. Ongoing and future preclinical and clinical trials will provide essential insights into vector design, disease correction at specific target organs, and the possibility of employing less toxic conditioning regimens to allow for the treatment and possible cure of LSDs through gene therapy approaches.
Conclusion
Because current therapeutic strategies are only partially successful in curing MPS, novel therapeutic approaches based on GT are being developed for various forms of these diseases. While preclinical studies have already demonstrated the potential benefit of both in-vivo and ex-vivo GT approaches in several MPS mouse models, phase I/II clinical trials are under development for various MPS. These strategies are being implemented with the rationale of providing high levels of the therapeutic enzyme to the patient either by direct infusion of the viral vector or by the engrafted gene-modified HSCs. This will hopefully allow for superior correction also at the level of organs that are more difficult to reach, such as the skeleton and the CNS. The development of strategies that allow us to overcome the BBB obstacle using in-vivo GT approaches with systemic injection is another challenge for the near future. The possibility of directly modifying defective genes by site-specific in-vivo genome editing is also being explored for a number of genetic diseases, including MPS.
The implementation of neonatal screening, which allows for early diagnosis, is of critical importance to ensure timely treatment of congenital disorders such as MPS IH in which a younger age at transplantation together with preservation of cognitive function have been reported as major predictors for superior cognitive development after allogeneic HSCT. Neonatal screening programs have already been developed in the US for SCID and some LSD, and pilot studies are on-going also in Italy in selected regions for ADA-SCID and MPS-IH [18, 65,66,67]. Thanks to the implementation of neonatal screening, and considering that UCB has become the preferential HSC source for allogeneic HSCT in LSD and in particular in MPS IH both in the US and Europe, UCB may also be considered in the future as an alternative source for HSC-GT in newborn/toddlers after dedicated storage of autologous UCB units. In this regard, strategies aimed at the expansion and improvement of transduction of primitive HSCs could expand the field of application of ex-vivo gene therapy [68]. The development of conditioning regimens associated with low extramedullary toxicity, such as those based on monoclonal antibodies selectively depleting blood cells in the BM [17], might open new frontiers to increase the application of HSC-GT at neonatal age.
Abbreviations
- AAV:
-
Adeno-associated virus
- BBB:
-
Blood–brain barrier
- BM:
-
Bone marrow
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- ERT:
-
Enzyme replacement therapy
- GAG:
-
Glycosaminoglycan
- GLD:
-
Globoid cell leukodystrophy
- GLP:
-
Good Laboratory Practice
- GT:
-
Gene therapy
- HIR:
-
Human insulin receptor
- HS:
-
Heparan sulfate
- HSC:
-
Hematopoietic stem cell
- HSCT:
-
Hematopoietic stem cell transplantation
- IDUA:
-
Alpha-l-iduronidase
- LDLR:
-
Low-density lipoprotein receptor
- LSD:
-
Lysosomal storage disease
- LV:
-
Lentivirus(al)
- mAb:
-
Monoclonal antibody
- MLD:
-
Metachromatic leukodystrophy
- MPS:
-
Mucopolysaccharidosi(e)s
- RV:
-
Retrovirus(al)
- SGSH:
-
N-sulfoglucosamine sulfohydrolase
- UCB:
-
Umbilical cord blood
- WT:
-
Wild-type
- ZFN:
-
Zinc finger nucleases
References
Neufeld EF, Muenzer J. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001.
Clarke LA. The mucopolysaccharidoses: a success of molecular medicine. Expert Rev Mol Med. 2008;10:e1.
Neufeld EF, Fratantoni JC. Inborn errors of mucopolysaccharide metabolism. Science. 1970;169:141–6.
Valayannopoulos V, Wijburg FA. Therapy for the mucopolysaccharidoses. Rheumatology (Oxford). 2011;50:v49–59.
Noh H, Lee JI. Current and potential therapeutic strategies for mucopolysaccharidoses. J Clin Pharm Ther. 2014;39:215–24.
Desnick RJ, Schuchman EH. Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu Rev Genomics Hum Genet. 2012;13:307–35.
Sanford M, Lo JH. Elosulfase alfa: first global approval. Drugs. 2014;74:713–8.
Fox JE, Volpe L, Bullaro J, Kakkis ED, Sly WS. First human treatment with investigational rhGUS enzyme replacement therapy in an advanced stage MPS VII patient. Mol Genet Metab. 2015;114:203–8.
Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111:63–72.
Hobbs JR, Hugh-Jones K, Barrett AJ, Byrom N, Chambers D, Henry K, et al. Reversal of clinical features of Hurler's disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet. 1981;2:709–12.
Aldenhoven M, Jones SA, Bonney D, Borrill RE, Coussons M, Mercer J, et al. Hematopoietic cell transplantation for mucopolysaccharidosis patients is safe and effective: results after implementation of international guidelines. Biol Blood Marrow Transplant. 2015;21:1106–9.
Aldenhoven M, Wynn RF, Orchard PJ, O'Meara A, Veys P, Fischer A, et al. Long-term outcome of hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. 2015;125:2164–72.
Staba SL, Escolar ML, Poe M, Kim Y, Martin PL, Szabolcs P, et al. Cord-blood transplants from unrelated donors in patients with Hurler's syndrome. N Engl J Med. 2004;350:1960–9.
Aldenhoven M, Boelens JJ, de Koning TJ. The clinical outcome of hurler syndrome after stem cell transplantation. Biol Blood Marrow Transplant. 2008;14:485–98.
Aldenhoven M, de Koning TJ, Wulffraat NM, Boelens JJ. Umbilical cord blood from an unrelated donor as source for stem cell transplantations in inborn errors of metabolism. Ned Tijdschr Geneeskd. 2008;152:1719–24.
Tomatsu S, Azario I, Sawamoto K, Pievani AS, Biondi A, Serafini M, et al. Neonatal cellular and gene therapies for mucopolysaccharidoses: the earlier the better? J Inherit Metab Dis. 2016;39:189–202.
Palchaudhuri R, Saez B, Hoggatt J, Schajnovitz A, Sykes DB, Tate TA, et al. Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nat Biotechnol. 2016;34:738–45.
Elliott S, Buroker CJJ, Potier AM, Trometer JD, Elbin C, et al. Pilot study of newborn screening for six lysosomal storage diseases using tandem mass spectrometry. Mol Genet Metab. 2016;118:304–9.
Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med. 2015;66:471–86.
Sands MS, Davidson BL. Gene therapy for lysosomal storage diseases. Mol Ther. 2006;13:839–49.
Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet. 2011;12:341–55.
Simonato M, Bennett J, Boulis NM, Castro MG, Fink DJ, Goins WF, et al. Progress in gene therapy for neurological disorders. Nat Rev Neurol. 2013;9:277–91.
Pardridge WM. Drug and gene delivery to the brain: the vascular route. Neuron. 2002;36:555–8.
Pardridge WM. Molecular biology of the blood-brain barrier. Mol Biotechnol. 2005;30:57–70.
Desmaris N, Verot L, Puech JP, Caillaud C, Vanier MT, Heard JM. Prevention of neuropathology in the mouse model of hurler syndrome. Ann Neurol. 2004;56:68–76.
Winner LK, Beard H, Hassiotis S, Lau AA, Luck AJ, Hopwood JJ, Hemsley KM. A preclinical study evaluating AAVrh10-based gene therapy for Sanfilippo syndrome. Hum Gene Ther. 2016;27:363–75.
Cressant A, Desmaris N, Verot L, Bréjot T, Froissart R, Vanier MT, et al. Improved behavior and neuropathology in the mouse model of Sanfilippo type IIIB disease after adeno-associated virus-mediated gene transfer in the striatum. J Neurosci. 2004;24:10229–39.
Liu G, Chen YH, He X, Martins I, Heth JA, Chiorini JA, Davidson BL. Adeno-associated virus type 5 reduces learning deficits and restores glutamate receptor subunit levels in MPS VII mice CNS. Mol Ther. 2007;15:242–7.
Tardieu M, Zerah M, Husson B, de Bournonville S, Deiva K, Adamsbaum C, et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: results of a phase I/II trial. Hum Gene Ther. 2014;25:506–16.
Wolf DA, Lenander AW, Nan Z, Belur LR, Whitley CB, Gupta P, et al. Direct gene transfer to the CNS prevents emergence of neurologic disease in a murine model of mucopolysaccharidosis type I. Neurobiol Dis. 2011;43:123–33.
Fraldi A, Hemsley K, Crawley A, Lombardi A, Lau A, Sutherland L, et al. Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum Molec Genet. 2007;16:2693–702.
Haurigot V, Marco S, Ribera A, Garcia M, Ruzo A, Villacampa P, et al. Whole body correction of mucopolysaccharidosis IIIA by intracerebrospinal fluid gene therapy. J Clin Invest.2013;123(8):3254–71.
Fu H, DiRosario J, Kang L, Muenzer J, McCarty DM. Restoration of central nervous system alpha-N-acetylglucosaminidase activity and therapeutic benefits in mucopolysaccharidosis IIIB mice by a single intracisternal recombinant adeno-associated viral type 2 vector delivery. J Gene Med. 2010;12:624–33.
Karolewski BA, Wolfe JH. Genetic correction of the fetal brain increases the lifespan of mice with the severe multisystemic disease mucopolysaccharidosis type VII. Mol Ther. 2006;14:14–24.
Sorrentino NC, Maffia V, Strollo S, Cacace V, Romagnoli N, Manfredi A, et al. A comprehensive map of CNS transduction by eight recombinant adeno-associated virus serotypes upon cerebrospinal fluid administration in pigs. Mol Ther. 2016;24:276–86.
Cotugno G, Annunziata P, Tessitore A, et al. Long-term amelioration of feline mucopolysaccharidosis VI after AAV-mediated liver gene transfer. Mol Ther. 2011;19:461–9.
Hinderer C, Bell P, Gurda BL, Wang Q, Louboutin JP, Zhu Y, et al. Liver-directed gene therapy corrects cardiovascular lesions in feline mucopolysaccharidosis type I. Proc Natl Acad Sci U S A. 2014;111:14894–9.
Brady RO, Schiffmann R. Enzyme-replacement therapy for metabolic storage disorders. Lancet Neurol. 2004;3:752–6.
Sorrentino NC, Fraldi A. Brain targeting in MPS-IIIA. Pediatr Endocrinol Rev. 2016;13(Suppl 1):630–8.
Pardridge WM. Targeted delivery of protein and gene medicines through the blood-brain barrier. Clin Pharmacol Ther. 2015;97:347–61.
Sorrentino NC, D'Orsi L, Sambri I, Nusco E, Monaco C. Spampanato C, et al. A highly secreted sulphamidase engineered to cross the blood-brain barrier corrects brain lesions of mice with mucopolysaccharidoses type IIIA. EMBO Mol Med. 2013;5:675–90.
Boado RJ, Lu JZ, Hui EK, Pardridge WM. Insulin receptor antibody-sulfamidase fusion protein penetrates the primate blood-brain barrier and reduces glycosoaminoglycans in Sanfilippo type a cells. Mol Pharm. 2014;11:2928–34.
Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotech. 2009;27:59–65.
Zhang H, Yang B, Mu X, Ahmed SS, Su Q, He R, et al. Several rAAV vectors efficiently cross the blood-brain barrier and transduce neurons and astrocytes in the neonatal mouse central nervous system. Mol Ther. 2011;19:1440–8.
Kaminski J, Frederickson R. Special issue on genome editing technologies for gene and cell therapy. Mol Ther. 2016;24:407–654.
Bernardo ME, Aiuti A. The role of conditioning in hematopoietic stem cell gene therapy. Hum Gene Ther. 2016;27:741–8.
Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP. Baricordi C,et al. lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151.
Cicalese MP, Ferrua F, Castagnaro L, Pajno R, Barzaghi F, Giannelli S, et al. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood. 2016;128:45–54.
Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158.
Rosenberg JB, Kaminsky SM, Aubourg P, Crystal RG, Sondhi D. Gene therapy for metachromatic leukodystrophy. J Neurosci Res. 2016;94:1169–79.
Cicalese MP, Aiuti A. Clinical applications of gene therapy for primary immunodeficiencies. Hum Gene Ther. 2015;26:210–9.
Naldini L, Trono D, Verma IM. Lentiviral vectors, two decades later. Science. 2016;353:1101–2.
Zheng Y, Rozengurt N, Ryazantsev S, Kohn DB, Satake N, Neufeld EF. Treatment of the mouse model of mucopolysaccharidosis I with retrovirally transduced bone marrow. Mol Genet Metab. 2003;79:233–44.
Wang D, Zhang W, Kalfa TA, Grabowski G, Davies S, Malik P, Pan D. Reprogramming erythroid cells for lysosomal enzyme production leads to visceral and CNS cross-correction in mice with hurler syndrome. Proc Natl Acad Sci U S A. 2009;106:19958–63.
Biffi A, Capotondo A, Fasano S, del Carro U, Marchesini S, Azuma H, et al. Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J Clin Invest. 2006;116:3070–82.
Biffi A, De Palma M, Quattrini A, Del Carro U, Amadio S, Visigalli I, et al. correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J Clin Invest 2004;113:1118–1129.
Gentner B, Visigalli I, Hiramatsu H, Lechman E, Ungari S, Giustacchini A, et al. Identification of hematopoietic stem cell-specific miRNAs enables gene therapy of globoid leukodystrophy. Science Transl Med. 2010;2:58ra84.
Wakabayashi T, Shimada Y, Akiyama K, Higuchi T, Fukuda T, Kobayashi H, et al. Hematopoietic stem cell gene therapy corrects neuropathic phenotype in murine model of mucopolysaccharidosis type II. Hum Gene Ther. 2015;26:357–66.
Sessa M, Lorioli L, Fumagalli F, Acquati S, Redaelli D, Baldoli C, et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet. 2016;388:476–87.
Visigalli I, Delai S, Politi LS, Di Domenico C, Cerri F, Mrak E, et al. Gene therapy augments the efficacy of hematopoietic cell transplantation and fully corrects mucopolysaccharidosis type I phenotype in the mouse model. Blood. 2010;116:5130–9.
Visigalli I, Delai S, Ferro F, Cecere F, Vezzoli M, Sanvito F, et al. Preclinical testing of the safety and tolerability of LV-mediated above normal alpha-L-iduronidase expression in murine and human hematopoietic cells using toxicology and biodistribution GLP studies. Hum Gene Ther. 2016;27:813–29.
Tomatsu S, Alméciga-Díaz CJ, Montaño AM, Yabe H, Tanaka A, Dung VC, et al. Therapies for the bone in mucopolysaccharidoses. Mol Genet Metab. 2015;114:94–109.
Gatto F, Redaelli D, Salvadè A, Marzorati S, Sacchetti B, Ferina C, et al. Hurler disease bone marrow stromal cells exhibit altered ability to support osteoclast formation. Stem Cells Dev. 2012;21:1466–77.
Sergijenko A, Langford-Smith A, Liao AY, Pickford CE, McDermott J, Nowinski G, et al. Myeloid/microglial driven autologous hematopoietic stem cell gene therapy corrects a neuronopathic lysosomal disease. Mol Ther. 2013;21:1938–49.
Hopkins PV, Campbell C, Klug T, Rogers S, Raburn-Miller J, Kiesling J. Lysosomal storage disorder screening implementation: findings from the first six months of full population pilot testing in Missouri. J Pediatr. 2015;166:172–7.
van der Spek J, Groenwold RH, van der Burg M, van Montfrans JM. TREC based newborn screening for severe combined immunodeficiency disease: a systematic review. J Clin Immunol. 2015;35:416–30.
Ombrone D, Giocaliere E, Forni G, Malvagia S, La Marca G. Expanded newborn screening by mass spectrometry: new tests, future perspectives. Mass Spectrom Rev. 2016;35:71–84.
Zonari E, Desantis G, Petrillo C, Boccalatte FE, Lidonnici MR, Kajaste-Rudnitski A, et al. Efficient ex vivo engineering and expansion of highly purified human hematopoietic stem and progenitor cell populations for gene therapy. Stem Cell Reports. 2017;8:977–90.
Funding
AA and BG have received a research grant from Telethon Foundation for MPS IH GT. MS, AF, and MEB are holders of research grants from Telethon Foundation. The publication costs for this paper in the IJP supplement were made possible with unconditional financial support from BioMarin, Sanofi Genzyme, and Shire. The sponsors had no input into the content of articles, which were independently prepared by the authors and have undergone the journal’s standard peer-review process.
Availability of data and materials
Data reported in this review are published in peer-reviewed journals or have been reported in presentations during scientific meetings.
About this supplement
This article has been published as part of Italian Journal of Pediatrics, Volume 44 Supplement 2, 2018: Mucopolysaccharidoses: state of the art. The full contents of the supplement are available online at https://ijponline.biomedcentral.com/articles/supplements/volume-44-supplement-2 .
Author information
Authors and Affiliations
Contributions
AF, MS, AA, and MEB coorganized the review structure and cowrote the manuscript; NCS and BG contributed to manuscript writing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
AA is the Principal Investigator of the TIGET-MLD clinical trial sponsored by GlaxoSmithKline (GSK). The remaining authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fraldi, A., Serafini, M., Sorrentino, N.C. et al. Gene therapy for mucopolysaccharidoses: in vivo and ex vivo approaches. Ital J Pediatr 44 (Suppl 2), 130 (2018). https://doi.org/10.1186/s13052-018-0565-y
Published:
DOI: https://doi.org/10.1186/s13052-018-0565-y