
Abstract
Pediatric refractory epilepsy is a broad phenotypic spectrum with great genetic heterogeneity. Next-generation sequencing (NGS) combined with Sanger sequencing could help to understand the genetic diversity and underlying disease mechanisms in pediatric epilepsy. Here, we report sequencing results from a cohort of 172 refractory epilepsy patients aged 0–14 years. The pathogenicity of identified variants was evaluated in accordance with the American College of Medical Genetics and Genomics (ACMG) criteria. We identified 43 pathogenic or likely pathogenic variants in 40 patients (23.3%). Among these variants, 74.4% mutations (32/43) were de novo and 60.5% mutations (26/43) were novel. Patients with onset age of seizures ≤12 months had higher yields of deleterious variants compared to those with onset age of seizures > 12 months (P = 0.006). Variants in ion channel genes accounted for the greatest functional gene category (55.8%), with SCN1A coming first (16/43). 81.25% (13/16) of SCN1A mutations were de novo and 68.8% (11/16) were novel in Dravet syndrome. Pathogenic or likely pathogenic variants were found in the KCNQ2, STXBP1, SCN2A genes in Ohtahara syndrome. Novel deleterious variants were also found in West syndrome, Doose syndrome and glucose transporter type 1 deficiency syndrome patients. One de novo MECP2 mutation were found in a Rett syndrome patient. TSC1/TSC2 variants were found in 60% patients with tuberous sclerosis complex patients. Other novel mutations detected in unclassified epilepsy patients involve the SCN8A, CACNA1A, GABRB3, GABRA1, IQSEC2, TSC1, VRK2, ATP1A2, PCDH19, SLC9A6 and CHD2 genes. Our study provides novel insights into the genetic origins of pediatric epilepsy and represents a starting-point for further investigations into the molecular pathophysiology of pediatric epilepsy that could eventually lead to better treatments.
Similar content being viewed by others
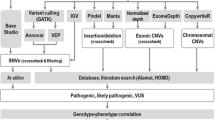


Introduction
Epilepsy is a complex group of chronic brain disorders that are characterized by recurrent spontaneous seizures, and these can often begin in childhood. Repeated and refractory seizures can cause long-term cognitive impairment, decreased social participation and significantly lower quality of life [1, 2]. Epilepsy is one of the most common neurological disorders with 50 to 100 million affected worldwide, and 2 to 4 million new cases diagnosed each year [3].
Epilepsy is a heterogeneous disease with diverse clinical manifestations and causes, including altered ion channel expression, neurotransmitter signaling, synaptic structure, gliosis, and inflammation [1]. Estimates of heritability from twin studies range from 25% to 70% [4, 5]. Although the range of heritability estimates is quite large, disparate studies using varied methods and studying divergent populations are all consistent in concluding that there is a substantial inherited component to epilepsy [6]. Because of this, we sought to investigate the genome in a heterogeneous set of patients with epilepsy and their parents, with the hope that we would identify novel mutations and confirm existing reports of genetic associations with epilepsy. This type of genetic information can provide an entry point into the biology of epilepsy that could eventually lead to new molecular treatment targets.
With the rapid progress of next-generation sequencing (NGS) techniques, our knowledge of the genetic etiology in many brain disorders such as epilepsy, autism and intellectual disability has expanded greatly [7, 8]. NGS is now capable of efficient and accurate sequencing of entire genomes with small amounts of tissue at ever decreasing costs and has required new approaches to analysing the very large amount of data obtained. For this study, our priority was to separate common and benign genetic variants from those that are likely to be related to the cause of epilepsy, and we chose to apply the American College of Medical Genetics and Genomics (ACMG) guidelines [9,10,11]. The ACMG guidelines classify variants into pathogenic, likely pathogenic, uncertain significance, likely benign, and benign categories based on genetic information that includes population, functional, computational and segregation data. In this study, we investigated 153 epilepsy candidate genes in a cohort of 172 refractory epilepsy pediatric patients. We aimed to provide genetic diagnoses of this patient cohort and explore the genetic etiology of pediatric refractory epilepsy.
Method
Participants
We retrospectively collected and analyzed 172 cases of pediatric refractory epilepsy patients between the ages of 1 day to 14 years old in the Department of Pediatrics of Qilu Hospital, China. The program adhered to guidelines of patients’ consent for participation and research was supported by the Ethics Committee of Qilu hospital, Shandong University (No. 2016(027)).
All patients were examined and diagnosed at the Pediatric Department in Qilu Hospital using a combination of patients’ illness history, previous history, family history, physical examinations, developmental evaluation, hematological examination, ambulatory or video electroencephalography (AEEG/VEEG) monitoring, magnetic resonance imaging (MRI) or computed tomography (CT), and genetic sequencing. Developmental evaluation included gross motor, fine motor, language, and personal-social skills. The above information was reviewed by two qualified pediatric epileptologists. Seizure types and epilepsy syndromes were diagnosed and classified according to the guidelines of International League Against Epilepsy (2014, 2017) [12, 13].
Next-generation sequencing
Targeted gene capture and sequencing
Blood samples of the patients and their biological parents were collected to test if the mutations were de novo or inherited. Genomic DNA was extracted from peripheral blood using the QIAamp DNA Mini Kit (Qiagen, China).
One hundred fifty-three genes (Table 1) associated with epilepsy were selected by a gene capture strategy, using the GenCap custom enrichment kit (MyGenostics, China) following the manufacturer’s protocol. The biotinylated capture probes were designed to tile all of the exons without repeated regions. The captured DNAs were eluted, amplified and then their polymerase chain reaction (PCR) products were purified with SPRI beads (Beckman, USA). The enriched libraries were sequenced for paired-end reads of 150 bp by Illumina HiSeq X Ten.
Data analysis and pathogenicity of candidate variants
After sequencing, raw data were saved in FASTQ format. Illumina sequencing adapters and low quality reads (< 80 bp) were filtered by Cutadapt [14]. Clean reads were aligned to UCSC hg19 human reference genome using the Burrows-Wheeler Alignment [15] tool. Duplicated reads were removed using Picard (http://broadinstitute.github.io/picard). Insertions, deletions and SNP variants were detected and filtered using the Genome Analysis Toolkit [16]. Then the identified variants were annotated using ANNOVAR [17] and associated with the following databases: 1000 genomes, Exome Aggregation Consortium, The Human Gene Mutation Database, and predicted by Mutation Taster (MT) [18], Sorting Intolerant From Tolerant (SIFT) [19], PolyPhen-2 (PP2) [20] and Genomic Evolutionary Rate Profiling (GERP++) [21, 22]. Splice-site were predicted by Human Splicing Finder [23]. All variants identified by the Illumina HiSeq X Ten sequencer were confirmed by Sanger sequencing. The pathogenicity of mutations was assessed in accordance with American College of Medical Genetics and Genomics guideline (ACMG) [9,10,11].
Statistical analysis
Statistical analysis was performed using SPSS19. The yields of deleterious variants in patients with different onset age or family history were compared using the chi-squared test.
Results
In the current study, we recruited 172 epilepsy pediatric patients, including 23 with Dravet syndrome, ten with Ohtahara syndrome, two with Ohtahara syndrome evolving to West syndrome, ten with West syndrome, two with West syndrome evolving to Lennox-Gastaut syndrome, five with Lennox-Gastaut syndrome, four with Doose syndrome, two with epilepsy of infancy with migrating focal seizures, two with epileptic encephalopathy with continuous spike and wave during sleep, and one each with temporal lobe epilepsy, early myoclonic encephalopathy, Landau-Kleffner syndrome, and glucose transporter type 1 deficiency syndrome. Three patients had Rett syndrome, five had tuberous sclerosis complex, and one had Sturge-Weber syndrome. Forty-two patients were diagnosed as unclassified epileptic encephalopathy and 57 patients were diagnosed as unclassified refractory epilepsy due to nonspecific manifestations (Table 2).
One hundred fifty-three epilepsy-related genes were selected for sequencing in all patients. The expression pattern of the targeted 153 genes across tissues were analyzed and classified according to the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov) and The Human Protein Atlas (https://www.proteinatlas.org) database (Additional file 1: Table S1). In our 153-gene panel, 51 genes show elevated expression, 14 genes have low expression, and 88 of them exhibit medium levels of expression in brain. The 14 low-expression genes have been associated with epilepsy, including: ARG1 [24,25,26,27], ARHGEF15 [28], CASR [29, 30], CHRNA2 [31], DBH [32,33,34], DIAPH3 [35], FOLR1 [36, 37], GABRA6 [38, 39], GLRA1 [40, 41], NID2 [42, 43], PROC [44], SLC13A5 [45, 46], SLC19A3 [47], SRPX2 [48]. Specifically, among 51 elevated genes in brain, 4 genes (GABRG2, GABBR2, GABRA1, GRIN1) show restricted brain expression.
The DNA samples of patients were analyzed by using NGS and the variants were validated by Sanger Sequencing. For the samples subjected to targeted sequencing, the quality assurance (QA) /quality control (QC) file are provided in Additional file 1: Table S2.
After sequencing the 153 epilepsy genes, we identified 43 deleterious variants in 23.3% patients (40 of 172), with three children harbouring more than one deleterious variant. Our results were similar to previous reports, with diagnostic yields ranging between 10% and 48.5% [49,50,51,52,53,54,55,56]. There were 60.5% (26/43) novel deleterious variants found in our study. A total of 43 variants in 22 genes were scored as pathogenic or likely pathogenic, including SCN1A (16), TSC2 (5), STXBP1 (2), SCN8A (2), TSC1(1), MECP2 (1), CHD2 (1), PCDH19 (1), GABRA1 (1), GABRB3 (1), SLC2A1 (1), SLC9A6 (1), IQSEC2 (1), KCNQ2 (1), SCN2A (1), CACNA1A (1), KCNT1 (1), SYNGAP1 (1), ATP1A2 (1), CDKL5 (1), ADSL (1), VRK2 (1) (Fig. 1a). Among these 43 pathogenic or likely pathogenic variants, there were 18 (41.9%) missense mutations, 3 (7%) splice site mutations, 11 (25.6%) nonsense mutations, 10 (23.3%) frame-shifts, and 1 (2.3%) deletion mutations (Fig. 1a, Table 3).

Mutated pathogenic or likely pathogenic genes in 172 refractory epilepsy children. a The frequency of mutated genes scored as pathogenic or likely pathogenic adhered to ACMG; b Functional classification of the mutated pathogenic or likely pathogenic genes; c The yield of pathogenic and likely pathogenic variants according to the electroclinical phenotype. Abbreviations: DS, Dravet syndrome; OS, Ohtahara syndrome; OS-WS, Ohtahara syndrome evolves to West syndrome; WS, West syndrome; WS-LGS, West syndrome evolves to Lennox-Gastaut syndrome; LGS, Lennox-Gastaut syndrome; Doose, Doose syndrome; ECSWS, epileptic encephalopathy with continuous spike and wave during sleep; EIMFS, epilepsy of infancy with migrating focal seizures; TLE, temporal lobe epilepsy; EME, early myoclonic encephalopathy; LKS, Landau-Kleffner syndromes; UEE, unclassified epileptic encephalopathy; GLUT1-DS, glucose transporter type 1 deficiency syndrome; Rett, Rett syndrome; TSC, tuberous sclerosis complex; SWS, Sturge-Weber syndrome; UE, unclassified refractory epilepsy
More recent studies suggest that many severe epilepsy types begin in infancy or childhood, especially those with psychomotor retardation and epileptic encephalopathies are often due to de novo mutations [30, 31]. In our study, 32/43 (74.4%) pathogenic or likely pathogenic variants were de novo, five (11.6%) were paternal, one (2.3%) was maternal, and five (11.6%) were unknown due to blood samples from parents were unavailable (Table 3).
To further explore the genetic pathogenesis of epilepsy, we subdivided the mutated genes into nine groups according to the molecular and biological function of the gene produce. These functional groups included voltage-gated ion channels, enzyme/enzyme modulators, membrane trafficking, ligand-gated ion channels, DNA/RNA binding, cell-adhesion proteins, glucose transporter, proton antiporter, and GTP/GDP exchanges. Variants in ion channel genes (SCN1A, SCN2A, SCN8A, CACNA1A, KCNT1, KCNQ2) accounted for 51.2% (22/43) of the pathogenic or likely pathogenic variants. Variants in enzyme/enzyme modulator genes (TSC1, TSC2, SYNGAP1, ATP1A2, CDKL5, ADSL, VRK2) accounted for 25.6% (11/43) of pathogenic or likely pathogenic variants. Variants in genes encoded membrane trafficking (STXBP1), ligand-gated ion channels (GABRA1, GABRB3), DNA/RNA binding proteins (MECP2, CHD2) each accounted for 4.7% (2/43) (Fig. 1b). Ion channels (voltage-gated and ligand-gated) accounted for 55.8% in total, suggesting that dysfunction of ion channels plays critical roles in the pathogenesis of epilepsy.
We then analyzed the yield of the epilepsy gene panel testing based on electroclinical syndrome (Fig. 1c). The yield of deleterious variants in Dravet syndrome (69.6%, 16/23) and glucose transporter type 1 deficiency syndrome (100%, 1/1) patients was higher than that in others. Patients with onset age of seizures ≤12 months had higher yields of deleterious variants compared to those with onset age of seizures > 12 months (31/101 vs 9/71; χ2 = 7.583, df = 1, P = 0.006). The family history did not affect whether or not a deleterious genetic variant was identified (7/27 vs 33/145; χ2 = 0.128, df = 1, P = 0.804).
There were 16 mutations in SCN1A gene, of which six (37.5%) were missense mutations, one (6.25%) was a splice site mutation, four (25%) were nonsense mutations, four (25%) were frame-shifts, and one (6.25%) was deletion mutation. Thirteen of the 16 (81.3%) SCN1A mutations were de novo and 11 (68.8%) were novel. We further analysed the positions of the mutations in the affected proteins corresponding to gene mutations and found that 43.8% (7/16) of protein changes are in the intracellular loop of sodium channel protein type 1 subunit alpha, 31.3% (5/16) are in the extracellular loop, 18.8% (3/16) are in the transmembrane region, and 6.25% (1/16) are in the pore forming area (Fig. 2).

Schematic representation of the mutations in subunit alpha of sodium channel type 1 (SCN1A) in our study. SCN1A alpha unit has four domains (I–IV), each domain includes 6 transmembrane segments (S1–S6). Purple circle = mutation; AEDs, anti-epileptic drugs. The position of mutations in SCN1A is approximate and is according to reference transcript NM_001165963.
There has been a marked increase in genetic diagnoses of a number of key childhood-onset epilepsy syndromes, such as Dravet syndrome, which has been mainly linked to SCN1A [17]. In our 16 patients diagnosed as Dravet syndrome with pathogenic or likely pathogenic variants, all identified mutations were in the SCN1A gene. These 16 Dravet syndrome patients had typical manifestations: onset between 3 to 8 months of age, fever-sensitive, multiple seizure types, and developmental delay after seizure onset. 81.25% (13/16) SCN1A mutations were de novo in Dravet syndrome patients and one was inherited from the father who had a history of febrile seizures (FS). 12.5% (2/16) SCN1A mutations were unknown. Pathogenic and likely pathogenic mutations each accounted for 50% (Table 4). 50% (8/16) of the Dravet syndrome variants cause nonsense or frameshift mutations that result in truncated proteins, which was consistent with a previous study [57]. We evaluated whether different seizure types, family history, abnormal brain MRI, or developmental delay were associated with specific SCN1A mutation types or locations within the gene. We did not detect any bias towards particular regions of the gene or in the type of mutation, although our small sample size did not provide substantial power (Additional file 1: Tables S3 and S4).
Twelve patients presented typical manifestation of Ohtahara syndrome: onset age within postnatal 30 days, tonic spasms, burst suppression EEG and developmental delay. Pathogenic or likely pathogenic variants in Ohtahara syndrome were in the KCNQ2 (1), STXBP1 (1), SCN2A (1) genes. The nonsense mutation in STXBP1 (c.364C > T, p.R122X) was detected in one Ohtahara syndrome patients that evolved to West syndrome. This patient had an onset age of postnatal 17 day, spasms, and burst-suppression EEG at postnatal 22 day and hypsarrhythmia EEG at 4 months (Table 5).
West syndrome patients in our study had onset ages of seizures ranging from postnatal 19 days to 6 months. Typical clinical manifestations were all observed, including spasms, hypsarrhythmia EEG, and developmental delay. 16.7% (2/12) of the West syndrome children evolves to Lennox-Gastaut syndrome. After sequencing, we identified 4 pathogenic or likely pathogenic mutations in the following genes: STXBP1 (1), KCNT1 (1), CDKL5 (1), ADSL (1). 75% (3/4) of these variants were de novo.
One of the West syndrome patients were found to carry two mutations: a nonsense ADSL (c.253C > T, p.R85X) mutation was scored as likely pathogenic and was inherited from her unaffected mother. Another reported missense ADSL (c.71C > T, p.P24L) [58] mutation which was inherited from her unaffected father were scored as uncertain pathogenicity. ADSL has been reported to be related to adenylosuccinate lyase deficiency, which is an autosomal recessive defect of purine metabolism [59, 60]. The patient presented with spasms 2 months after birth. Brain MRI showed cerebral dysplasia and EEG showed hypsarrhythmia and multifocal discharges. The patient also had developmental delay and lack of eye contact. A definitive diagnosis can be made with high performance liquid chromatography examination of the urine to detect the ratio of succinyladenosine and succinyl-aminoimidazole carboximide riboside, but this was not available for the patient in question. Thus, this patient was diagnosed clinically as having West syndrome.
A novel frame-shift mutation in SYNGAP5 (c.274_277del, p.G92fs) was detected in a patient with Doose syndrome. This patient presented with myoclonic and myoclonic-astatic seizures, as well as having atypical absence seizures. SYNGAP5 had been reported to be associated with Doose syndrome and mental retardation, autosomal dominant 5 (MRD5) [51, 61, 62]. This mutation, which is very rare, was de novo, and caused frameshift changes in Ras/Rap GTPase-activating protein SynGAP, was therefore scored as pathogenic (Table 5).
One glucose transporter type 1 deficiency syndrome patient presented with seizures at age 28 months. The patient has alopecia and was almost bald at 4 years old. The child did not have other abnormalities in blood tests, brain MRI, or neurological exam. Her cerebrospinal fluid glucose value was 2.04 mmol/L (blood glucose value was 7.2 mmol/L before lumbar puncture; fasting blood glucose value was 5.2 mmol/L). NGS identified a missense mutation in SLC2A1 (c.296T > G, p.M99R). The mutation was de novo and novel. The patient’s parents and sister were normal, which is consistent with the sequencing results. Symptoms improved with a ketogenic diet, with seizures controlled for more than 6 months.
One MECP2 mutation (c.799C > T, p.R267X) was detected in a girl diagnosed as Rett syndrome. The girl developed normally for the first 18 months, gradually lost speech ability while developing repetitive hand-wringing. Seizures began at age 3 years. The MECP2 gene is located on the X-chromosome, and Rett syndrome is inherited through this gene in a dominant fashion [63]. This patient had a de novo MECP2 nonsense mutation, consistent with her parents being unaffected.
40% (2/5) of tuberous sclerosis complex patients were diagnosed with West syndrome associated with tuberous sclerosis complex in our study. Tuberous sclerosis complex is closely related to the TSC1/TSC2 genes [64,65,66,67].
In our study, all of the tuberous sclerosis complex patients’ initial presentations were seizures, of which 80% (4/5) presented in the first year of life. 60% (3/5) had hypomelanotic macules and 40% (2/5) had multi nodules. One patient’s only clinical manifestation was seizures and three (60%) patients with seizures had only one major feature of tuberous sclerosis complex. After sequencing, 60% (3/5) patients were found to have deleterious TSC1 or TSC2 mutations.
We identified more than one TSC1/2 mutations in 2 patients. One patient has two TSC2 mutations inherited from his affected father. Facial angiofibromas appeared by age 3–4 years in 60% (3/5) patients in the follow-up period. Gilboa et al. [68] reported four patients with the same TSC1 genomic deletion (9q34.13q34.2) in a family and none of them fulfilled the clinical criteria for tuberous sclerosis complex. In our study, one patient with pathogenic TSC1 (c.2768_2769insC, p.L924Ffs*26) mutation presented with focal seizures beginning at age four. There were two hypopigmented macules on the patient’s abdomen. The brain MRI results were normal and there are no other features of tuberous sclerosis complex. This de novo mutation causes a frame-shift in hamartin and has not been reported previously. Thus, this patient was considered to have unclassified refractory epilepsy.
One unclassified epileptic encephalopathy patient had two deleterious mutations: SCN8A inherited from his affected father (c.4324G > A, p.E1442K) and IQSEC2 (c.4246_4247insG, p.S1416fs). Early-infantile epileptic encephalopathies (EIEE) caused by SCN8A mutations are designated as EIEE13 (OMIM #614558) [69]. The missense mutation in SCN8A is very rare in the general population, and had been previously predicted to be damaging by SIFT, MT and PP2. IQSEC2 is an X-linked gene that has been reported to be related to intellectual disability and epilepsy, and it encodes the IQ motif and SEC7 domain-containing protein 2 [70]. The identified novel IQSEC2 mutation was de novo and was scored as being pathogenic.
Other pathogenic or likely pathogenic mutations found in patients with unclassified epileptic encephalopathy included CACNA1A, GABRA1, GABRB3, PCDH19, and CHD2. Epileptic encephalopathies with the above mutations had been designated as EIEE42, EIEE19, EIEE43, EIEE9 and EEOC (childhood-onset epileptic encephalopathy) according to Online Mendelian Inheritance in Man (OMIM). Other deleterious variants found in patients with unclassified refractory epilepsy were in VRK2, ATP1A2, and SLC9A6. Taking these unclassified epileptic encephalopathies and unclassified refractory epilepsy patients’ clinical manifestations into consideration, we found that all patients with deleterious mutations in genes encoding ion channels (SCN8A, CACNA1A, GABRB3, GABRA1) had similar clinical symptoms: onset age of seizures within the first year, epileptic encephalopathy and developmental delay. In contrast, patients with mutations in VRK2, ATP1A2, and SLC9A6, had relatively later onset age of seizures.
We then assessed the clinical benefit of genetic testing in those patients with identified deleterious variants. NGS helped with the diagnosis (n = 8), medication selection (n = 18), reproductive planning (n = 4), and treatment planning (n = 1). The finding of the SLC2A1 variant in Case 164 prompted other tests such as cerebrospinal fluid (CSF) glucose that were clinically useful. Identification of deleterious SCN1A mutations in five young infants with clinically suspected Dravet syndrome helped early diagnosis (Case 13, 38, 65, 115, 140) and led to the discontinuation of oxcarbazepine (Case 13) that exacerbated seizures. Identification of SCN1A mutations in other Dravet syndrome patients helped to avoid sodium channel blockers such as oxcarbazepine, carbamazepine and lamotrigine. Among the four Dravet syndrome patients who responded to anticonvulsants (Case 13, 26, 149, 172), 75% (3/4) of them were prescribed sodium valproate or clonazepam suggesting that these medications may be effective in Dravet syndrome. The finding of the TSC2 variants in Cases 94 and 98 helped early diagnosis and Case 32 experienced remission with administration of rapamune. Identification of TSC1 prompted clinical surveillance for tuberous sclerosis complex in Case 68. The findings of patients with deleterious variants in TSC2 (Case 32, 98), SCN8A (Case 7), SCN1A (Case 149), ADSL (Case 52) which were inherited, helped in prenatal counselling (Table 6).
Discussion
Epilepsy is highly heterogeneous and can be primarily genetic in origin, or be secondary to structural or metabolic disorders of the central nervous system [71, 72]. To date, over 500 genes have been implicated in epilepsy [73,74,75,76]. However, the overlapping clinical features of different epilepsy syndromes and non-specific phenotypes can hamper clinical and genetic diagnosis [53]. The correct genetic diagnosis can help to guide treatment and prognosis. In addition to genetic origins, pediatric epilepsy may also arise from epigenetic mechanisms mediating gene-environment interactions during neurodevelopment. In this study, we used NGS to investigate 153 epilepsy related genes in a cohort of 172 refractory epilepsy children.
Approximately one quarter of genes identified in epilepsy encode ion channel proteins, including voltage-gated channels (Na+, K+, Ca2+ channels and hyperpolarization-activated cyclic nucleotide-gated channels) and ligand-gated ion channels (N-Methyl-D-Aspartate receptors, Gamma-aminobutyric acid receptors and Nicotinic Acetylcholine receptors) [77]. The genes that encode ion channels and are relevant to epilepsy include SCN1A, SCN1B, SCN2A, SCN8A, KCNA1, KCNA2, KCNB1, KCNC1, KCNMA1, KCNQ2, KCNQ3, KCNT1, KCTD7, HCN1, CACAN1A, CACNA1H, GRIN1, GRIN2A, GRIN2B, GRIN2D, GABRA1, GABRB3, GABRG2, CHRNA2, CHRNA4, CHRNB2. In our study, 51.2% pathogenic or likely pathogenic variants were found in voltage-gated ion channels and 4.7% were found in ligand-gated ion channels. Thus, we further confirmed that ion channels play an important role in the pathogenesis of epilepsy.
An SCN1A mutation was first discovered in epilepsy in 2000 [72], and now hundreds of new SCN1A mutations have been described in epilepsy patients, making it the most common epilepsy-related gene [78]. In our study, we found SCN1A mutations in 16/44 deleterious variants, making it the most common gene to show variation in our study. SCN1A encodes the Nav1.1 pore-forming α-subunit, expressed mainly in inhibitory GABAergic neurons. The α-subunit comprises four homologous domains (I–IV), forming a tetrameric structure. Each domain is composed of six transmembrane segments (S1–S6) [77]. The S4, voltage-sensing segment has multiple positively charged amino acids. The intracellular loop between III and IV domain functions as the inactivation gate. The α-subunit is usually associated with two β-subunits that influence α-subunit localization and function [77]. Among α-subunit of sodium channel genetic variants in our study, 43.8% (7/16) are within the intracellular loop, 31.3% (5/16) in the extracellular loop, 18.8% (3/16) in the transmembrane area, and 6.25% (1/16) in the pore forming area. All the extracellular mutations are between S5 and S6, which is very close to the pore forming area. These variants may influence the initiation and propagation of action potentials, making these inhibitory GABAergic neurons less excitable. Some antiepileptic drugs (AEDs) bind to the inner cavity of the pore of the sodium channel (IS6, IIIS6 and IVS6) [77, 79]. The pore forming area or internal/external loop could be promising targets for new seizure prophylaxis medications.
Patients harboring SCN1A mutations can have with Dravet syndrome or generalized epilepsy with febrile seizures plus. One Dravet syndrome patient inherited the SCN1A mutations from his father only had febrile seizures. This could be due to somatic mosaicism [72, 80, 81]. A Dravet syndrome mouse model (Nav1.1 knockout-based) responded well to stiripentol and clobazam, which are commonly used to treat Dravet syndrome [82,83,84,85]. One of the patients in our study was treated with oxcarbazepine, which blocks sodium channels and worsened seizures, before the diagnosis of Dravet syndrome was made. This case illustrates the importance of correct molecular diagnosis in selecting the best anticonvulsant.
Approximately half of Ohtahara syndrome patients with STXBP1 mutations evolve to West syndrome [86]. In our study, there was one such patient with a nonsense mutation in STXBP1, suggesting that this gene could play a role in the etiology of West syndrome. Our findings also suggest that STXBP1 is related to both Ohtahara syndrome and West syndrome.
KCNT1 is associated with epilepsy of infancy with migrating focal seizures, autosomal dominant nocturnal frontal lobe epilepsy, and other types of early onset epileptic encephalopathies [87,88,89]. Ohba et al. [88] found 11 KCNT1 mutations in a total of 362 epilepsy patients: 9/18 epilepsy of infancy with migrating focal seizures cases (50%), 1/180 West syndrome cases (0.56%), and 1/66 unclassified early onset epileptic encephalopathy cases (1.52%), suggesting that KCNT1 may be a causal gene for West syndrome. In our study, one KCNT1 (c.862G > A, p.G288S) mutation was found in a patient diagnosed as West syndrome.
Genetic studies of neuropsychiatric disease have led to the discovery of molecular etiology and pathophysiology. For example, most cases of Rett syndrome are now known to arise from mutations in the MECP2 gene, which codes for a methyl-CpG-binding protein 2 [90]. Another example is glucose transporter type 1 deficiency syndrome, which has been attributed to variants in SLC2A1, SLC2A2, and GLUT1. In our study, the glucose transporter type 1 deficiency syndrome patient did not have cerebrospinal fluid analysis as part of their diagnostic work-up until the genetic data suggested the diagnosis. This example illustrates the utility of NGS in clinical scenarios, and in time this may become an important part of the evaluation of pediatric patients with epilepsy. In some epilepsy syndromes, crucial interventions such as diet modification can have dramatic beneficial effects, so early diagnosis is vital [91, 92].
In our study, SCN1A was the main deleterious variant in Dravet syndrome and KCNQ2, STXBP1, SCN2A were found in Ohtahara syndrome. Deleterious variants in STXBP1, KCNT1, CDKL5, ADSL genes were found in West syndrome. Novel mutations in SYNGAP1 were found in Doose syndrome, a SLC2A1 mutation was found in GLUT1-DS and a de novo MECP2 mutation were found in Rett syndrome. TSC1/TSC2 variants were found in 60% of patients with tuberous sclerosis complex. Mutations found in unclassified epileptic encephalopathy were mainly in ion-channel genes. Thus, our study reinforces previous observations that the clinical syndrome and genetic etiology do not always match.
We tested 153 epilepsy genes and found 43 pathogenic and likely pathogenic variants in this study. Considering that over 500 epilepsy genes have been reported [73,74,75,76], our work was not comprehensive, which is a limitation of this study. With the decreasing cost of whole genome sequencing, the interrogation of the entire genome is now feasible for larger samples of epilepsy patients, and this approach has already been fruitful in other neuropsychiatric disorders such as autism, Kabuki syndrome, Bohring-Opitz syndrome and others [93, 94].
For genetic testing, it is proposed to conduct the strong candidate gene sequencing first (SCN1A for Dravet syndrome, MECP2 for Rett syndrome and TSC1/2 for tuberous sclerosis complex) before a NGS multi-gene panel testing [95,96,97]. In our study, we conducted targeted panel sequencing on Dravet syndrome and Rett syndrome patients before screening the strongest candidate gene for the following reasons. First, the correct clinical diagnosis of these syndromes can be difficult, especially in some of the younger patients in our sample, and often requires longitudinal assessment, which delays the correct diagnosis. Thus, we elected to perform NGS on our subjects before knowing the clinical diagnosis in some cases, such as these syndromes. Since our NGS panel that contains 153 epilepsy genes, our approach could facilitate the correct diagnosis in some cases. Second, it is now apparent that while 70–80% Dravet syndrome patients have SCN1A mutations, mutations in other genes such as SCN1B, SCN2A, SCN8A, PCDH19, GABRA1, GABRG2, STXBP1, CHD2 genes can cause Dravet syndrome like phenotypes [98], which would be missed if only SCN1A was sequenced. Similarly, CDKL5 and FOXG1 have been associated with atypical Rett syndrome [99], in addition to MECP2.
In tuberous sclerosis complex patients, we have a similar clinical scenario in which most features of tuberous sclerosis complex become evident only after 3 years of age, limiting their usefulness for early diagnosis [100]. In our study, all of the tuberous sclerosis complex patients’ initial presentations were seizures, of which 80% presented in the first year of life. 60% had hypomelanotic macules and 40% had multi nodules. 20% patient’s only clinical manifestation was seizures and 60% patients with seizures had only one major feature of tuberous sclerosis complex. 60% patients were found to have deleterious TSC1 or TSC2 mutations by NGS sequencing. Facial angiofibromas appeared by age 3–4 years in 60% patients in the follow-up period.
In summary, we identified 43 pathogenic or likely pathogenic variants, of which 26 mutations were novel and 32 were de novo. Variants in ion channel genes accounted for the largest category of gene in children with refractory epilepsy. Dravet syndrome is closely related to the SCN1A gene, which was the most frequently-appearing gene showing variants in our study. Novel and de novo mutations were found in Ohtahara syndrome, West syndrome, Doose syndrome and tuberous sclerosis complex pediatric patients. We also found a novel mutation in glucose transporter type 1 deficiency. Our results reinforce the importance and feasibility of precise genetic diagnosis for epilepsy, with the hope that in future, this will both aid in understanding the molecular pathophysiology and lead to new treatment targets.
Change history
16 October 2018
Following publication of the original article [1], the authors reported that one of the authors’ names is spelled incorrectly.
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- AEDs:
-
antiepileptic drugs
- EEOC:
-
childhood-onset epileptic encephalopathy
- EIEE:
-
early-infantile epileptic encephalopathies
- MT:
-
Mutation Taster
- NGS:
-
next-generation sequencing
- OMIM:
-
Online Mendelian Inheritance in Man
- PP2:
-
PolyPhen-2
- SCN1A:
-
subunit alpha of sodium channel type 1
- SIFT:
-
Sorting Intolerant From Tolerant
References
Henshall DC, Hamer HM, Pasterkamp RJ, Goldstein DB, Kjems J, Prehn JHM, et al. MicroRNAs in epilepsy: pathophysiology and clinical utility. Lancet Neurol. 2016;15(13):1368–76.
Nickels KC, Zaccariello MJ, Hamiwka LD, Wirrell EC. Cognitive and neurodevelopmental comorbidities in paediatric epilepsy. Nat Rev Neurol. 2016;12(8):465–76.
Pitkanen A, Loscher W, Vezzani A, Becker AJ, Simonato M, Lukasiuk K, et al. Advances in the development of biomarkers for epilepsy. Lancet Neurol. 2016;15(8):843–56.
Miller LL, Pellock JM, DeLorenzo RJ, Meyer JM, Corey LA. Univariate genetic analyses of epilepsy and seizures in a population-based twin study: the Virginia Twin Registry. Genet Epidemiol. 1998;15(1):33–49.
Kjeldsen MJ, Kyvik KO, Christensen K, Friis ML. Genetic and environmental factors in epilepsy: a population-based study of 11900 Danish twin pairs. Epilepsy Res. 2001;44(2–3):167–78.
Speed D, O'Brien TJ, Palotie A, Shkura K, Marson AG, Balding DJ, et al. Describing the genetic architecture of epilepsy through heritability analysis. Brain. 2014;137:2680–9.
Brandler WM, Sebat J. From de novo mutations to personalized therapeutic interventions in autism. Annu Rev Med. 2015;66:487–507.
Hoischen A, Krumm N, Eichler EE. Prioritization of neurodevelopmental disease genes by discovery of new mutations. Nat Neurosci. 2014;17(6):764–72.
Directors ABo. ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing. Genet Med. 2015;17(1):68–9.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15(7):565–74.
Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55(4):475–82.
Fisher RS, Cross JH, D'Souza C, French JA, Haut SR, Higurashi N, et al. Instruction manual for the ILAE 2017 operational classification of seizure types. Epilepsia. 2017;58(4):531–42.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011; https://doi.org/10.14806/ej.17.1.200.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–33.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–2.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Cooper GM, Stone EA, Asimenos G, Program NCS, Green ED, Batzoglou S, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15(7):901–13.
Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol. 2010;6(12):e1001025.
Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9):e67.
Uchino T, Haraguchi Y, Aparicio JM, Mizutani N, Higashikawa M, Naitoh H, et al. Three novel mutations in the liver-type arginase gene in three unrelated Japanese patients with argininemia. Am J Hum Genet. 1992;51(6):1406–12.
Carvalho DR, Brand GD, Brum JM, Takata RI, Speck-Martins CE, Pratesi R. Analysis of novel ARG1 mutations causing hyperargininemia and correlation with arginase I activity in erythrocytes. Gene. 2012;509(1):124–30.
Uchino T, Snyderman SE, Lambert M, Qureshi IA, Shapira SK, Sansaricq C, et al. Molecular basis of phenotypic variation in patients with argininemia. Hum Genet. 1995;96(3):255–60.
Wu TF, Liu YP, Li XY, Wang Q, Ding Y, Ma YY, et al. Five novel mutations in ARG1 gene in Chinese patients of argininemia. Pediatr Neurol. 2013;49(2):119–23.
Veeramah KR, Johnstone L, Karafet TM, Wolf D, Sprissler R, Salogiannis J, et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia. 2013;54(7):1270–81.
Kapoor A, Satishchandra P, Ratnapriya R, Reddy R, Kadandale J, Shankar SK, et al. An idiopathic epilepsy syndrome linked to 3q13.3-q21 and missense mutations in the extracellular calcium sensing receptor gene. Ann Neurol. 2008;64(2):158–67.
Sato K, Hasegawa Y, Nakae J, Nanao K, Takahashi I, Tajima T, et al. Hydrochlorothiazide effectively reduces urinary calcium excretion in two Japanese patients with gain-of-function mutations of the calcium-sensing receptor gene. J Clin Endocrinol Metab. 2002;87(7):3068–73.
Kurahashi H, Hirose S. Autosomal Dominant Nocturnal Frontal Lobe Epilepsy. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews((R)). Seattle (WA): University of Washington, Seattle; 1993–2018.
Lauterborn JC, Ribak CE. Differences in dopamine beta-hydroxylase immunoreactivity between the brains of genetically epilepsy-prone and Sprague-Dawley rats. Epilepsy Res. 1989;4(3):161–76.
Schank JR, Liles LC, Weinshenker D. Reduced anticonvulsant efficacy of valproic acid in dopamine beta-hydroxylase knockout mice. Epilepsy Res. 2005;65(1–2):23–31.
Warter JM, Coquillat G, Kurtz D. Human circulating dopamine-beta-hydroxylase and epilepsy. Psychopharmacologia. 1975;41(1):75–9.
Lesca G, Rudolf G, Labalme A, Hirsch E, Arzimanoglou A, Genton P, et al. Epileptic encephalopathies of the Landau-Kleffner and continuous spike and waves during slow-wave sleep types: genomic dissection makes the link with autism. Epilepsia. 2012;53(9):1526–38.
Steele SU, Cheah SM, Veerapandiyan A, Gallentine W, Smith EC, Mikati MA. Electroencephalographic and seizure manifestations in two patients with folate receptor autoimmune antibody-mediated primary cerebral folate deficiency. Epilepsy Behav. 2012;24(4):507–12.
Perez-Duenas B, Toma C, Ormazabal A, Muchart J, Sanmarti F, Bombau G, et al. Progressive ataxia and myoclonic epilepsy in a patient with a homozygous mutation in the FOLR1 gene. J Inherit Metab Dis. 2010;33(6):795–802.
Kumari R, Lakhan R, Kalita J, Garg RK, Misra UK, Mittal B. Potential role of GABAA receptor subunit; GABRA6, GABRB2 and GABRR2 gene polymorphisms in epilepsy susceptibility and pharmacotherapy in North Indian population. Clin Chim Acta. 2011;412(13–14):1244–8.
Hernandez CC, Gurba KN, Hu N, Macdonald RL. The GABRA6 mutation, R46W, associated with childhood absence epilepsy, alters 6beta22 and 6beta2 GABA(A) receptor channel gating and expression. J Physiol. 2011;589(Pt 23):5857–78.
Elmslie F, Gardiner M. Genetics of the epilepsies. Curr Opin Neurol. 1995;8(2):126–9.
Bakker MJ, van Dijk JG, van den Maagdenberg AMJM, Tijssen MAJ. Startle syndromes. Lancet Neurol. 2006;5(6):513–24.
Conroy J, McGettigan PA, McCreary D, Shah N, Collins K, Parry-Fielder B, et al. Towards the identification of a genetic basis for Landau-Kleffner syndrome. Epilepsia. 2014;55(6):858–65.
Dong L, Chen Y, Lewis M, Hsieh JC, Reing J, Chaillet JR, et al. Neurologic defects and selective disruption of basement membranes in mice lacking entactin-1/nidogen-1. Lab Investig. 2002;82(12):1617–30.
Fong CY, Mumford AD, Likeman MJ, Jardine PE. Cerebral palsy in siblings caused by compound heterozygous mutations in the gene encoding protein C. Dev Med Child Neurol. 2010;52(5):489–93.
Thevenon J, Milh M, Feillet F, St-Onge J, Duffourd Y, Juge C, et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am J Hum Genet. 2014;95(1):113–20.
Hardies K, de Kovel CG, Weckhuysen S, Asselbergh B, Geuens T, Deconinck T, et al. Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain. 2015;138(Pt 11):3238–50.
Zeng WQ, Al-Yamani E, Acierno JS Jr, Slaugenhaupt S, Gillis T, MacDonald ME, et al. Biotin-responsive basal ganglia disease maps to 2q36.3 and is due to mutations in SLC19A3. Am J Hum Genet. 2005;77(1):16–26.
Roll P, Rudolf G, Pereira S, Royer B, Scheffer IE, Massacrier A, et al. SRPX2 mutations in disorders of language cortex and cognition. Hum Mol Genet. 2006;15(7):1195–207.
Hildebrand MS, Myers CT, Carvill GL, Regan BM, Damiano JA, Mullen SA, et al. A targeted resequencing gene panel for focal epilepsy. Nerology. 2016;86(17):1605–12.
Parrini E, Marini C, Mei D, Galuppi A, Cellini E, Pucatti D, et al. Diagnostic targeted resequencing in 349 patients with drug-resistant pediatric epilepsies identifies causative mutations in 30 different genes. Hum Mutat. 2017;38(2):216–25.
Carvill GL, Heavin SB, Yendle SC, McMahon JM, O'Roak BJ, Cook J, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45(7):825–30.
Trump N, McTague A, Brittain H, Papandreou A, Meyer E, Ngoh A, et al. Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis. J Med Genet. 2016;53(5):310–7.
Lemke JR, Riesch E, Scheurenbrand T, Schubach M, Wilhelm C, Steiner I, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia. 2012;53(8):1387–98.
Møller RS, Larsen LH, Johannesen KM, Talvik I, Talvik T, Vaher U, et al. Gene panel testing in epileptic encephalopathies and familial epilepsies. Mol Syndromol. 2016;7(4):210–9.
Kodera H, Kato M, Nord AS, Walsh T, Lee M, Yamanaka G, et al. Targeted capture and sequencing for detection of mutations causing early onset epileptic encephalopathy. Epilepsia. 2013;54(7):1262–9.
Ortega-Moreno L, Giráldez BG, Soto-Insuga V, Losada-Del Pozo R, Rodrigo-Moreno M, Alarcón-Morcillo C, et al. Molecular diagnosis of patients with epilepsy and developmental delay using a customized panel of epilepsy genes. PLoS One. 2017;12(11):e0188978. https://doi.org/10.1371/journal.pone.0188978.
Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest. 2005;115(8):2010–7.
Mao X, Li K, Tang B, Luo Y, Ding D, Zhao Y, et al. Novel mutations in ADSL for Adenylosuccinate Lyase deficiency identified by the combination of trio-WES and constantly updated guidelines. Sci Rep. 2017;7(1):1625.
Jurecka A, Zikanova M, Kmoch S, Tylki-Szymańska A. Adenylosuccinate lyase deficiency. J Inherit Metab Dis. 2015;38(2):231–42.
Chen BC, McGown IN, Thong MK, Pitt J, Yunus ZM, Khoo TB, et al. Adenylosuccinate lyase deficiency in a Malaysian patient, with novel adenylosuccinate lyase gene mutations. J Inherit Metab Dis. 2010;33(Suppl 3):S159–62.
Hamdan FF, Gauthier J, Spiegelman D, Noreau A, Yang Y, Pellerin S, et al. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med. 2009;360(6):599–605.
Berryer MH, Hamdan FF, Klitten LL, Møller RS, Carmant L, Schwartzentruber J, et al. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum Mutat. 2013;34(2):385–94.
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8.
Hasbani DM, Crino PB. Tuberous sclerosis complex. Handb Clin Neurol. 2018;148:813–22.
Zhang H, Nanba E, Yamamoto T, Ninomiya H, Ohno K, Mizuguchi M, et al. Mutational analysis of TSC1 and TSC2 genes in Japanese patients with tuberous sclerosis complex. J Hum Genet. 1999;44(6):391–6.
Wilson PJ, Ramesh V, Kristiansen A, Bove C, Jozwiak S, Kwiatkowski DJ, et al. Novel mutations detected in the TSC2 gene from both sporadic and familial TSC patients. Hum Mol Genet. 1996;5(2):249–56.
Hoogeveen-Westerveld M, Wentink M, van den Heuvel D, Mozaffari M, Ekong R, Povey S, et al. Functional assessment of variants in the TSC1 and TSC2 genes identified in individuals with tuberous sclerosis complex. Hum Mutat. 2011;32(4):424–35.
Gilboa T, Segel R, Zeligson S, Alterescu G, Ben-Pazi H. Ganglioglioma, epilepsy, and intellectual impairment due to familial TSC1 deletion. J Child Neurol. 2018;33(7):482–6.
Wagnon JL, Meisler MH. Recurrent and non-recurrent mutations of SCN8A in epileptic encephalopathy. Front Neurol. 2015;6(104) https://doi.org/10.3389/fneur.2015.00104.
Zerem A, Haginoya K, Lev D, Blumkin L, Kivity S, Linder I, et al. The molecular and phenotypic spectrum of IQSEC2-related epilepsy. Epilepsia. 2016;57(11):1858–69.
Wilmshurst JM, Berg AT, Lagae L, Newton CR, Cross JH. The challenges and innovations for therapy in children with epilepsy. Nat Rev Neurol. 2014;10(5):249–60.
Oyrer J, Maljevic S, Scheffer IE, Berkovic SF, Petrou S, Reid CA. Ion channels in genetic epilepsy: from genes and mechanisms to disease-targeted therapies. Pharmacol Rev. 2018;70(1):142–73.
Orsini A, Zara F, Striano P. Recent advances in epilepsy genetics. Neurosci Lett. 2018;667:4–9.
Weber YG, Biskup S, Helbig KL, Von Spiczak S, Lerche H. The role of genetic testing in epilepsy diagnosis and management. Expert Rev Mol Diagn. 2017;17(8):739–50.
Wang J, Lin ZJ, Liu L, Xu HQ, Shi YW, Yi YH, et al. Epilepsy-associated genes. Seizure. 2017;44:11–20.
Samarasinghe TD, Sands SA, Skuza EM, Joshi MS, Nold-Petry CA, Berger PJ. The effect of prenatal maternal infection on respiratory function in mouse offspring: evidence for enhanced chemosensitivity. J Appl Physiol (1985). 2015;119(3):299–307.
Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57(4):397–409.
Oliva M, Berkovic SF, Petrou S. Sodium channels and the neurobiology of epilepsy. Epilepsia. 2012;53(11):1849–59.
McDonald CL, Saneto RP, Carmant L, Sotero de Menezes MA. Focal seizures in patients with SCN1A mutations. J Child Neurol. 2017;32(2):170–6.
Depienne C, Trouillard O, Gourfinkel-An I, Saint-Martin C, Bouteiller D, Graber D, et al. Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. J Med Genet. 2010;47(6):404–10.
Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74(1):41–8.
Hawkins NA, Anderson LL, Gertler TS, Laux L, George AL Jr, Kearney JA. Screening of conventional anticonvulsants in a genetic mouse model of epilepsy. Ann Clin Transl Neurol. 2017;4(5):326–39.
Chiron C, Dulac O. The pharmacologic treatment of Dravet syndrome. Epilepsia. 2011;52(Suppl 2):72–5.
Cao D, Ohtani H, Ogiwara I, Ohtani S, Takahashi Y, Yamakawa K, et al. Efficacy of stiripentol in hyperthermia-induced seizures in a mouse model of Dravet syndrome. Epilepsia. 2012;53(7):1140–5.
Oakley JC, Cho AR, Cheah CS, Scheuer T, Catterall WA. Synergistic GABA-enhancing therapy against seizures in a mouse model of Dravet syndrome. J Pharmacol Exp Ther. 2013;345(2):215–24.
Di Meglio C, Lesca G, Villeneuve N, Lacoste C, Abidi A, Cacciagli P, et al. Epileptic patients with de novo STXBP1 mutations: key clinical features based on 24 cases. Epilepsia. 2015;56(12):1931–40.
Ko A, Youn SE, Kim SH, Lee JS, Kim S, Choi JR, et al. Targeted gene panel and genotype-phenotype correlation in children with developmental and epileptic encephalopathy. Epilepsy Res. 2018;141:48–55.
Ohba C, Kato M, Takahashi N, Osaka H, Shiihara T, Tohyama J, et al. De novo KCNT1 mutations in early-onset epileptic encephalopathy. Epilepsia. 2015;56(9):e121–8.
Fukuoka M, Kuki I, Kawawaki H, Okazaki S, Kim K, Hattori Y, et al. Quinidine therapy for West syndrome with KCNTI mutation: a case report. Brain Dev. 2017;39(1):80–3.
Leonard H, Cobb S, Downs J. Clinical and biological progress over 50 years in Rett syndrome. Nat Rev Neurol. 2017;13(1):37–51.
Zhou P, He N, Zhang JW, Lin ZJ, Wang J, Yan LM, et al. Novel mutations and phenotypes of epilepsy-associated genes in epileptic encephalopathies. Genes Brain Behav. 2018. https://doi.org/10.1111/gbb.12456
Daci A, Bozalija A, Jashari F, Krasniqi S. Individualizing treatment approaches for epileptic patients with glucose transporter type1 (GLUT-1) deficiency. Int J Mol Sci. 2018;19(1):122.
Ku CS, Polychronakos C, Tan EK, Naidoo N, Pawitan Y, Roukos DH, et al. A new paradigm emerges from the study of de novo mutations in the context of neurodevelopmental disease. Mol Psychiatry. 2013;18(2):141–53.
Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13(8):565–75.
Hirose S, Scheffer IE, Marini C, De Jonghe P, Andermann E, Goldman AM, et al. SCN1A testing for epilepsy: application in clinical practice. Epilepsia. 2013;54(5):946–52.
Kammoun F. Screening of MECP2 coding sequence in patients with phenotypes of decreasing likelihood for Rett syndrome: a cohort of 171 cases. J Med Genet. 2004;41(6):e85.
Avgeris S, Fostira F, Vagena A, Ninios Y, Delimitsou A, Vodicka R, et al. Mutational analysis of TSC1 and TSC2 genes in tuberous sclerosis complex patients from Greece. Sci Rep. 2017;7(1):16697.
Steel D, Symonds JD, Zuberi SM, Brunklaus A. Dravet syndrome and its mimics: beyond SCN1A. Epilepsia. 2017;58(11):1807–16.
Guerrini R, Parrini E. Epilepsy in Rett syndrome, and CDKL5- and FOXG1-gene-related encephalopathies. Epilepsia. 2012;53(12):2067–78.
Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372(9639):657–68.
Depienne C, Trouillard O, Saint-Martin C, Gourfinkel-An I, Bouteiller D, Carpentier W, et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet. 2009;46(3):183–91.
Nabbout R, Gennaro E, Dalla Bernardina B, Dulac O, Madia F, Bertini E, et al. Spectrum of SCN1A mutations in severe myoclonic epilepsy of infancy. Neurology. 2003;60(12):1961–7.
Harkin LA, McMahon JM, Iona X, Dibbens L, Pelekanos JT, Zuberi SM, et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain. 2007;130(Pt 3):843–52.
Fukuma G, Oguni H, Shirasaka Y, Watanabe K, Miyajima T, Yasumoto S, et al. Mutations of neuronal voltage-gated Na+ channel alpha 1 subunit gene SCN1A in core severe myoclonic epilepsy in infancy (SMEI) and in borderline SMEI (SMEB). Epilepsia. 2004;45(2):140–8.
Saitsu H, Kato M, Koide A, Goto T, Fujita T, Nishiyama K, et al. Whole exome sequencing identifies KCNQ2 mutations in Ohtahara syndrome. Ann Neurol. 2012;72(2):298–300.
Ishii A, Shioda M, Okumura A, Kidokoro H, Sakauchi M, Shimada S, et al. A recurrent KCNT1 mutation in two sporadic cases with malignant migrating partial seizures in infancy. Gene. 2013;531(2):467–71.
Mayer K, Ballhausen W, Rott HD. Mutation screening of the entire coding regions of the TSC1 and the TSC2 gene with the protein truncation test (PTT) identifies frequent splicing defects. Hum Mutat. 1999;14:401–11.
Gardella E, Becker F, Møller RS, Schubert J, Lemke JR, Larsen LH, et al. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann Neurol. 2016;79(3):428–36.
Epi4K Consortium. De novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am J Hum Genet. 2016;99(2):287–98.
Larsen J, Carvill GL, Gardella E, Kluger G, Schmiedel G, Barisic N, et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology. 2015;84(5):480–9.
Hirouchi M, Suzuki H, Itoda M, Ozawa S, Sawada J-i, Ieiri I, et al. Characterization of the cellular localization, expression level, and function of SNP variants of MRP2/ABCC2. Pharm Res. 2004;21(5):742–8.
Fokstuen S, Makrythanasis P, Hammar E, Guipponi M, Ranza E, Varvagiannis K, et al. Experience of a multidisciplinary task force with exome sequencing for Mendelian disorders. Hum Genomics. 2016;10(1):24.
Acknowledgements
All authors greatly appreciate to the families that take part in this research.
Funding
The project was funded by National Key Research and Development Program of China (NO. 2016YFC1306202), Key Research and Development Plan in Shandong Province (NO. 2016GSF201073), and General program of Qilu hospital, Shandong University (NO.2015QLMS08).
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
BML and JL were responsible for the original concept and the overall design of the research. JL, LLT, BML, RPS analyzed the EEG results and diagnosed patients. JL, SSS, YN, JL, XW, FL collected the clinical data and sample. JL, LLT, SSS, YN, XW, JL, JZ, FL, JW carried the experiments and analysed the sequencing data. JL, HYL performed structural and functional analysis experiments. JL, BML, CZ, AW, FL wrote and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The program adhered to guidelines of patients’ consent for participation and research and was supported by the Ethics Committee of Qilu hospital, Shandong University (No. 2016(027)).
Consent for publication
All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional information
The original version of this article was revised: the name of one of the authors had been spelled incorrectly. It should be Clement C. Zai, not Celement C. Zai.
Additional file
Additional file 1:
Table S1. The expression levels of the 153 targeted genes in brain. Table S2. The quality assurance (QA) /quality control (QC) of targeted sequencing. Table S3. The frequencies of different mutation locations in SCN1A gene and their corresponding phenotypes in Dravet syndrome patients. Table S4. The frequencies of different mutation types in SCN1A gene and their corresponding phenotypes in Dravet syndrome patients. (DOCX 98 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, J., Tong, L., Song, S. et al. Novel and de novo mutations in pediatric refractory epilepsy. Mol Brain 11, 48 (2018). https://doi.org/10.1186/s13041-018-0392-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-018-0392-5