
Abstract
Background
Machine learning methods and conventions are increasingly employed for the analysis of large, complex biomedical data sets, including genome-wide association studies (GWAS). Reproducibility of machine learning analyses of GWAS can be hampered by biological and statistical factors, particularly so for the investigation of non-additive genetic interactions. Application of traditional cross validation to a GWAS data set may result in poor consistency between the training and testing data set splits due to an imbalance of the interaction genotypes relative to the data as a whole. We propose a new cross validation method, proportional instance cross validation (PICV), that preserves the original distribution of an independent variable when splitting the data set into training and testing partitions.
Results
We apply PICV to simulated GWAS data with epistatic interactions of varying minor allele frequencies and prevalences and compare performance to that of a traditional cross validation procedure in which individuals are randomly allocated to training and testing partitions. Sensitivity and positive predictive value are significantly improved across all tested scenarios for PICV compared to traditional cross validation. We also apply PICV to GWAS data from a study of primary open-angle glaucoma to investigate a previously-reported interaction, which fails to significantly replicate; PICV however improves the consistency of testing and training results.
Conclusions
Application of traditional machine learning procedures to biomedical data may require modifications to better suit intrinsic characteristics of the data, such as the potential for highly imbalanced genotype distributions in the case of epistasis detection. The reproducibility of genetic interaction findings can be improved by considering this variable imbalance in cross validation implementation, such as with PICV. This approach may be extended to problems in other domains in which imbalanced variable distributions are a concern.
Similar content being viewed by others


Background
Genome-wide association studies (GWAS) have been frequently critiqued for failing to explain the “missing heritability” of complex disease in terms of single-locus main effects [1, 2]. In addition to interrogating the contributions of rare variants, non-coding regions, structural variation, etc., a logical reactionary paradigm to embrace involves revisiting heritability estimates to consider the effect of interactions and developing approaches that acknowledge that loci do not exist in isolation but rather act in complex networks of interacting partners in the dynamic, three-dimensional genome and in tissue-specific and environmental context [3,4,5,6]. Utilizing pre-existing GWAS data to test a curated set of potentially biologically-relevant interactions, such as those identified as being plausible via expert knowledge, integrating data from gene set enrichment analyses, chromatin capture experiments, co-expression data sets, etc. provides a way to overcome the multiple testing burden of naively testing every possible interaction and motivates future bench science experimentation [7, 8]. Accordingly, machine learning methods are appealing for the analysis of this big, complex data, and have been applied to diverse problems and data types across the biological sciences [9, 10]. However, machine learning should not be viewed as a panacea that can be readily applied to all genomics problems. Beyond concerns regarding model choice and interpretability, there are numerous reasons why valid biological interactions may fail to appear statistically significant and vice versa [11,12,13]. Therefore, typical machine learning tools, techniques, and standards from other fields may need tweaking to be appropriate for use in genomics considering the unique biases in generating genomic data sets, the structure of the genome, the validity of model assumptions, etc.
Improving the reproducibility of machine learning analyses of genomic data will require methodological and analytic advances from not only both the computational and wet laboratory sides, but also their consideration in conjunction with each other as a greater whole. Sharing data publicly for secondary analyses, writing open-source code in executable notebook format, and using container and cloud services all contribute to a culture of reproducibility that enhances the capacity for integrative and innovative computational analyses [14,15,16,17]. Likewise, thoughtfully interrogating methodological, environmental, and other determinants of inconsistencies in bench experimentation results lends robustness to findings, and this greater understanding of sources of variation can in itself lead to worthwhile new hypotheses [18]. Ideally, technological supports such as mobile applications for data collection will increasingly allow for recording more complete and consistent data in a format that can be seamlessly analyzed with software tools developed or modified to consider the unique intricacies of the data at hand [19].
Epistasis, or the non-additive interaction between genotypes to produce phenotype, is difficult to detect statistically but is of biological interest in light of a multifactorial view of disease [20,21,22]. This study is motivated by poor cross-validation performance observed for epistasis data sets with an interaction between two single nucleotide polymorphisms (SNPs). A given SNP may be represented as a categorical variable with possible values of 0, 1, or 2 corresponding to doses of the minor allele, and by extension the interaction of two SNPs may be represented in terms of 9 categories reflecting the identities of SNP1 and SNP2. Internal cross validation is a widely-used standard for evaluating the performance of a machine learning analysis in which the data is split into two mutually exclusive partitions, a model is fit using the ‘training’ set, and its performance is evaluated on predicting the classes of the held out set of observations (the ‘testing’ or ‘validation’ set; not to be confused with an external independent replication data set which may also be referred to as ‘testing’ or ‘validation’) [23]. Typically the overall data set is split such that the resultant training and testing partitions are random, independent draws from the same probability distribution, although there are also methods that consider the data structure, generally in terms of maintaining outcome class proportions between the training and testing data sets [24,25,26]. In this study, we propose a new cross validation method, proportional instance cross validation (PICV), that preserves the relative distribution of an independent variable (in our example application, SNP-SNP interaction genotypes) when dividing the overall data set into train and test partitions. We demonstrate significantly improved sensitivity and positive predictive value across all tested scenarios with application of PICV relative to a traditional cross validation implementation. We additionally apply PICV to primary open-angle glaucoma GWAS data to investigate an interaction previously reported to be significant in two independent data sets. Although this interaction is not observed to be significant in our analysis, PICV produced more consistent estimates than a traditional cross validation implementation. This approach is not only appropriate for epistasis data but may be readily applied to comparable imbalanced variable problems.
Methods
Data set generation
All data sets were generated using GAMETES, a tool that produces epistatic models between SNPs and creates data sets based off these models [27]. Penetrance functions were generated for SNP-SNP interaction scenarios for all 15 combinations of minor allele frequencies (MAFs) of {0.5, 0.4, 0.3, 0.2, and 0.1}, with SNP heritability kept constant at 0.005 and population prevalences of 0.5, 0.1, and 0.02 (Table 1, Additional file 1: Tables S1 and S2). Although a prevalence of 0.5 may seem high for a given disease, numerous risk factors for chronic and complex diseases in the United States population that may be phenotypes of interest are as or more prevalent, including being overweight or obese, lack of physical activity, excessive sodium consumption, lack of fruit and vegetable consumption, etc. [28]. The simulated data with prevalence of 0.1 is intended to reflect the US prevalence of common complex diseases such as diabetes or cardiovascular disease [29]. The simulated data sets of 0.02 prevalence approximately reflect the US prevalence of primary open-angle glaucoma, which is investigated in the real data application [30]. Balanced case-control ratio data sets of size 2000 and 10,000 were generated for the 0.5 prevalence scenario and of size 10,000 for the 0.1 and 0.02 prevalence scenarios.
Minor allele frequencies and penetrance tables used to generate balanced case-control ratio data sets of size 2000 and 10,000. Heritability = 0.005 and prevalence = 0.5 constant across all simulations.
Implementation and evaluation of traditional cross validation
For each of the 15 scenarios for each investigated prevalence and sample size combination, we perform 1000 replicates of a standard cross validation procedure in which two-thirds of observations are randomly allocated to be used for training and the remaining third is used for testing. The training data is then used to fit the following logistic regression models with and without the SNP-SNP interaction:
Where P(case) is a binary indicator of case-control status, SNP1 and SNP2 are categorical variables in which 0 corresponds to the homozygous dominant genotype, 1 to the heterozygous, and 2 to the homozygous recessive, and SNP1*SNP2 corresponds to the Cartesian product of the two {00, 01, 02, 10, 11, 12, 20, 21, 22}.
These models fit to the training data are then used to predict case-control status for the held-out testing data, using a cutoff of 0.5 for case versus control prediction assignment from the fitted values. These predictions are then used to calculate the sensitivity, specificity, positive predictive value, and negative predictive value for the testing data.
Implementation and evaluation of proportional instance cross validation (PICV)
For the proportional instance cross validation procedure, rather than randomly allocating each observation to be included in the training or testing set, observations are allocated in a genotype-specific fashion (Fig. 1). Two-thirds of the observations of each SNP-SNP genotype are randomly allocated to be used for training and the remaining third is used for testing. Therefore the same total proportion of individuals used for training versus testing is maintained as in the traditional cross validation procedure, and additionally, the relative proportions of each genotype are preserved between the overall data set and the training and testing partitions. Model fitting with the training data, testing data predictions, and performance measure calculations are conducted as for the traditional cross validation.

Comparing traditional cross validation and proportional instance cross validation (PICV). a The overall distribution of 9 SNP-SNP interaction genotypes (the 9 categories that result from the interaction of two SNPs in a hypothetical population of individuals. Note: only one possible allocation is depicted. b Traditional cross validation in which 2/3 of observations are randomly allocated to the training set and the remaining 1/3 are allocated to the testing set can result in draws with imbalanced genotype proportions. c PICV randomly allocates 2/3 of observations of each genotype to the training set and the remaining 1/3 to the testing set, ensuring that the relative proportions of genotypes are maintained
Comparison of traditional cross validation and proportional instance cross validation (PICV)
For both traditional cross validation and PICV, we calculate the absolute value of the difference between training and testing for each of four performance measures (sensitivity, specificity, positive predictive value, and negative predictive value) over 1000 trials for each of the 15 scenarios. We calculate p-values for the two-sample Kolmogorov-Smirnov test with the null hypothesis that there is no difference between the traditional cross validation implementation and PICV distributions of the difference between training and testing for each performance measure, with the one-sided alternative that the PICV distribution is smaller, with a significance threshold of α = 0.05.
Results
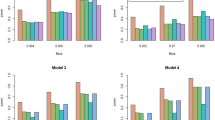
Implementing PICV for our simulated epistasis examples (that is, performing cross validation data set splitting such that observations are allocated to maintain the same relative proportions of each SNP-SNP genotype in the training and testing sets as in the data set overall) significantly improved the consistency between training and testing sensitivities and positive predictive values. Consistency between the training and testing data set performance measures is of interest as the PICV method addresses the discordance between training and testing partitions that can occur in traditional cross validation. Fig. 2 illustrates comparisons of training/testing consistencies for PICV versus a traditional cross validation procedure in which observations are allocated to the training and testing sets without regard to genotype (see Additional file 1: Figures S1-S60 for all minor allele frequency, prevalence, and cohort size combinations). P-values listed are for the two-sample Kolmogorov-Smirnov test of the distributions of the absolute values of the differences between the training and testing performance measure (e.g. sensitivity) over 1000 trials per scenario for these two cross validation approaches, with a one-sided alternative hypothesis that the split-by-genotype distribution is smaller. Table 2 summarizes these performance measures across all 15 SNP-SNP genotype MAF combination scenarios for the 0.5 prevalence simulations of size 2000 (see Additional file 1: Table S3 for prevalence = 0.5 and n = 10,000, Additional file 1: Table S4 for prevalence = 0.1, Additional file 1: Table S5 for prevalence = 0.02). Sensitivity and positive predictive value were significantly more consistent between test and train for PICV than for traditional cross validation across all 15 scenarios tested for both n = 2000 and n = 10,000. Although the specificity and negative predictive value comparisons mostly did not meet statistical significance, smaller medians and maximum values for the differences in these performance measures between training and testing were observed for the PICV approach for the majority of scenarios (Table 3). These results demonstrate that PICV is comparable to traditional cross validation in terms of specificity and negative predictive value while providing advantages in sensitivity and positive predictive value.

Consistency of training and testing performance measures for models with and without the interaction term, comparing a traditional cross validation procedure to PICV. Experimental scenario in which both SNPs have a MAF of 0.5, n = 2000. PPV: positive predictive value, NPV: negative predictive value, N.S.: not significant
Primary open-angle glaucoma interaction analysis
Prior interaction analyses of primary open-angle glaucoma identified several pairs of replicating interactions using the eMERGE and NEIGHBOR data [31]. We attempted to replicate the most significant interaction (between ALX4 and RBFOX1) in the GLAUGEN data set (dbGaP Study Accession: phs000308.v1.p1, available at https://www.ncbi.nlm.nih.gov/gap), which is harmonized with NEIGHBOR. The GLAUGEN model is adjusted for age, sex, site, and the first 6 principal components to reflect the eMERGE and NEIGHBOR models (the eMERGE and NEIGHBOR models additionally adjusted for platform, but all GLAUGEN samples were assessed on the same platform). Our analysis did not find a significant interaction between the two variants (Table 4). However, application of PICV to this data did yield training and testing p-values (0.376 and 0.323, respectively) more consistent with the overall LRT p-value (0.327) than a traditional cross validation procedure (0.442 and 0.470, respectively).
Discussion
Implementing a cross validation splitting procedure that maintains the relative proportions of each SNP-SNP genotype when dividing the overall data set significantly improved the sensitivity and positive predictive value consistencies between the training and testing partitions in each of the experimental scenarios tested. Although specificity and negative predictive value improvement did not meet statistical significance in most cases, application of the PICV approach did yield smaller median and maximum absolute differences between training and testing in the majority of scenarios. The interaction analysis did not replicate the prior finding between ALX4 and RBFOX1, however PICV still produced more consistent estimates than a traditional cross validation procedure for this data. Verma et al. note that RBFOX1 has been previously shown to be associated with myopia, and that eMERGE primary open-angle glaucoma cases had not been screened for myopia; GLAUGEN excluded individuals with more than 8 diopters of myopia. This inconsistent finding highlights the importance of considering epidemiological confounders and co-morbidities of complex phenotypes in genetic analyses.
Class imbalance is a well-recognized issue in machine learning analyses, particularly for the analysis of high-dimensional data sets as in genomics and other biomedical applications [32]. If the main objective of a machine learning analysis is maximizing accuracy, and the minority class is very small, simply predicting the majority class for each observation may yield high overall accuracy, as in the spam filtering problem [33]. Clearly, adoption of a balanced accuracy measure or a cost-sensitivity analysis that weighs the relative importance of avoiding false positives versus false negatives is critical for such problems, and numerous methods have been developed to address this issue including novel fitness functions, sampling-based approaches, and ensemble methods, including for epistasis modeling [34,35,36,37]. The present study, though thematically similar to the class imbalance problem, instead addresses imbalance in observations of classes of an independent variable, e.g. the SNP-SNP interaction genotype. This is also adjacent to the covariate and data set shift problems, in which the training and testing distributions differ (for example due to model training using clean data from consistent laboratory conditions to produce models that then fail to hold for experimentally gathered data with unanticipated environmental differences), but for internal cross validation [38,39,40]. Solutions to problems of both of these genres include re-weighting and –sampling techniques, whereas the present study circumvents the need for either via splitting the data to ensure balanced proportions by genotype between training and testing sets. The example application of imbalanced SNP-SNP genotypes considers a categorical variable, but the underlying idea of preserving the distribution of instances between training and testing with regard to an independent variable could be extended to continuous variables or combinations of variables via binning, propensity scores, etc.
Conclusions
Although the contribution of epistatic interactions may help explain the “missing heritability” of complex disease, statistical detection of epistasis remains challenging and can require adjustment of general machine learning protocols. With decreasing minor allele frequencies, the number of observations for rare SNP-SNP interaction genotypes becomes quite small in a GWAS of typical size, and a standard cross validation procedure may result in training/testing data set splits that poorly represent the data as a whole. This diminishes the ability to identify interactions of potential interest for experimental follow-up, and underscores the need to perform interaction analyses in an interaction-specific framework. A potentially overlooked element of performing reproducible analyses includes the imperative to develop and modify methods considering how intrinsic characteristics of the data and its structure may contribute to statistical failure to replicate despite biological (or other scientific) validity. Genomics and the biomedical sciences in general benefit from their increasingly multidisciplinary nature by incorporating methodology and theory from adjacent computational fields, but thoughtful contextualization of the data in view of the underlying biology is necessary to reap the potential benefits of applied machine learning methods and to successfully reproduce them.
References
Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747.
Visscher PM, et al. Five years of GWAS discovery. Am J Hum Genet. 2012;90(1):7–24.
Zuk O, et al. The mystery of missing heritability: genetic interactions create phantom heritability. P Natl Acad Sci. 2012;109(4):1193–8.
Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–93.
GTEx Consortium. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348(6235):648–60.
Cornelis MC, et al. The gene, environment association studies consortium (GENEVA): maximizing the knowledge obtained from GWAS by collaboration across studies of multiple conditions. Genet Epidemiol. 2010;34(4):364–72.
Bush WS, Dudek SM, Ritchie MD. Biofilter: a knowledge-integration system for the multi-locus analysis of genome-wide association studies. Pac Symp Biocomput. NIH Public Access. 2009;
Ritchie MD, et al. Methods of integrating data to uncover genotype-phenotype interactions. Nat Rev Gen. 2015;16(2):85.
Libbrecht MW, Noble WS. Machine learning in genetics and genomics. Nat Rev Gen. 2015;16(6):321.
Larranaga P, et al. Machine learning in bioinformatics. Brief Bioinform. 2006;7:86–112.
Moore JH, Williams SM. Traversing the conceptual divide between biological and statistical epistasis: systems biology and a more modern synthesis. BioEssays. 2005;27(6):637–46.
Greene CS, et al. Failure to replicate a genetic association may provide important clues about genetic architecture. PLoS One. 2009;4(6):e5639.
Moore JH, Asselbergs FW, Williams SM. Bioinformatics challenges for genome-wide association studies. Bioinformatics. 2010;26(4):445–55.
Longo DL, Drazen JM. Data sharing. N Engl J Med. 2016;7:276–7.
Kluyver T, et al. Jupyter notebooks-a publishing format for reproducible computational workflows: ELPUB; 2016. http://ebooks.iospress.nl/publication/42900.
Merkel D. Docker: lightweight linux containers for consistent development and deployment. Linux Journal. 2014;239(2014):2.
Beaulieu-Jones BK, Greene CS. Reproducibility of computational workflows is automated using continuous analysis. Nat Biotechnol. 2017;35(4):342–6.
Hines WC, et al. Sorting out the FACS: a devil in the details. Cell Rep. 2014;6(5):779–81.
Lithgow GJ, Driscoll M, Phillips P. A long journey to reproducible results. Nature News. 2017;548(7668):387.
Moore JH. The ubiquitous nature of epistasis in determining susceptibility to common human diseases. Hum Hered. 2003;56(1-3):73–82.
Carlborg Ö, Haley CS. Epistasis: too often neglected in complex trait studies? Nat Rev Gen. 2004;5(8):618.
Marchini J, Donnelly P, Cardon LR. Genome-wide strategies for detecting multiple loci that influence complex diseases. Nat Genet. 2005;37(4):413.
Arlot S, Celisse A. A survey of cross-validation procedures for model selection. Stat Surv. 2010;4:40–79.
van Hulse J, Khoshgoftaar TM, Napolitano A. Experimental perspectives on learning from imbalanced data. In: Proceedings of the 24th international conference on machine learning: ACM; 2007. http://ebooks.iospress.nl/publication/42900.
He H, Garcia EA. Learning from imbalanced data. IEEE T Knowl Data En. 2009;21(9):1263–84.
Estabrooks A, Jo T, Japkowicz N. A multiple resampling method for learning from imbalanced data sets. Comput Intell. 2004;20(1):18–36.
Urbanowicz RJ, et al. GAMETES: a fast, direct algorithm for generating pure, strict, epistatic models with random architectures. BioDat Min. 2012;5(1):16.
Centers for Disease Control and Prevention. Chronic Disease Prevention and Health Promotion. https://www.cdc.gov/chronicdisease/overview/index.htm. Accessed 05 Sep 2017.
National Center for Health Statistics. (2017). Heart Disease. Retrieved December 19, 2017, from https://www.cdc.gov/nchs/fastats/heart-disease.htm
Friedman DS, Wolfs RCW, O’Colmain BJ, Klein BE, Taylor HR, West S, et al. Prevalence of open-angle glaucoma among adults in the United States. Arch Ophthalmol (Chicago, Ill : 1960). 2004;122(4):532–8. https://doi.org/10.1001/archopht.122.4.532
Verma SS, Cooke Bailey JN, Lucas A, Bradford Y, Linneman JG, Hauser MA, et al. Epistatic gene-based interaction analyses for Glaucoma in eMERGE and NEIGHBOR consortium. PLoS Genet. 2016;12(9) https://doi.org/10.1371/journal.pgen.1006186
Lin W-J, Chen JJ. Class-imbalanced classifiers for high-dimensional data. Brief Bioinform. 2012;14(1):13–26.
Guzella TS, Caminhas WM. A review of machine learning approaches to spam filtering. Expert Syst Appl. 2009;36(7):10206–22.
Galar M, et al. A review on ensembles for the class imbalance problem: bagging-, boosting-, and hybrid-based approaches. IEEE T Syst Man Cyb C. 2012;42(4):463–84.
Liu X-Y, Wu J, Zhou Z-H. Exploratory undersampling for class-imbalance learning. IEEE T Syst Man Cyb B. 2009;39(2):539–50.
López V, et al. An insight into classification with imbalanced data: empirical results and current trends on using data intrinsic characteristics. Inform Sci. 2013;250:113–41.
Velez DR, et al. A balanced accuracy function for epistasis modeling in imbalanced datasets using multifactor dimensionality reduction. Genet Epi. 2007;31(4):306–15.
Quionero-Candela J, et al. Dataset shift in machine learning: The MIT Press; 2009. http://ebooks.iospress.nl/publication/42900.
Sugiyama M, et al. Direct importance estimation with model selection and its application to covariate shift adaptation. Adv Neur In Process Syst. 2008;20:1433–40.
Shimodaira H. Improving predictive inference under covariate shift by weighting the log-likelihood function. J Stat Plan Infer. 2000;90(2):227–44.
Funding
This work was supported by National Institutes of Health grants LM010098, LM12601, AI116794, DK112217, and ES013508.
Availability of data and materials
All data sets generated for this study are included in the Additional file 1 accompanying this article.
Author information
Authors and Affiliations
Contributions
JHM and ERP jointly conceptualized the proportional instance cross validation procedure. ERP simulated and analyzed the data. Both authors contributed to and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Supplemental tables and figures of data set simulation parameters and performance measures for all minor allele frequency, prevalence, and sample size combinations. (DOCX 3350 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Piette, E.R., Moore, J.H. Improving machine learning reproducibility in genetic association studies with proportional instance cross validation (PICV). BioData Mining 11, 6 (2018). https://doi.org/10.1186/s13040-018-0167-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13040-018-0167-7