
Abstract
Background
Male infertility is an increasing medical concern worldwide. In most cases, genetic factors are considered as the main cause of the disease. Globozoospermia (MIM102530) (also known as round-headed sperm) is a rare and severe malformed spermatospermia caused by acrosome deficiency or severe malformation. A subset of genetic mutations, such as DNAH6, SPATA16, DPY19L2, PICK1, and CCIN related to globozoospermia, have been reported in the past few years. The DPY19L2 mutation is commonly found in patients with globozoospermia. Herein, a 180-kbp homozygote deletion at 12q14.2 (g.63950001–64130000) was identified by copy number variation sequencing (CNVseq) in a patient with a globozoospermia, including the complete deletion of DPY19L2.
Case presentation
A 27-year-old patient at the First Affiliated Hospital of Xiamen University was diagnosed with infertility because, despite normal sexual activity for 4 years, his wife did not conceive. The patient was in good health with no obvious discomfort, no history of adverse chemical exposure, and no vices, such as smoking and drinking. The physical examination revealed normal genital development. However, semen tests showed a normal sperm count of 0% and the morphology was the round head. Sperm cytology showed that acrosomal enzyme was lower than normal. Reproductive hormones were in the normal range. B ultrasound did not show any abnormal seminal vesicle, prostate, bilateral testis, epididymis, and spermatic veins. The karyotype was normal, 46, XY, and no microdeletion of Y chromosome was detected. However, a homozygous deletion mutation was found in DPY19L2, which was further diagnosed as globozoospermia.
Conclusions
The present study reported a male infertility patient who was diagnosed with globozoospermia. The analysis of gene mutations revealed that DPY19L2 had a homozygous mutation, which was the primary cause of globozoospermia.
Similar content being viewed by others


Background
Infertility has gradually become a medical issue attracting worldwide attention. In most cases, the gene factors are considered to be a major cause of the disease [1, 2]. Globozoospermia (MIM102530) is a rare (incidence 0.1%) form of severe monomorphic teratozoospermia that leads to primary male infertility and is characterized by round-headed spermatozoa without acrosome, an abnormal nuclear membrane, and midpiece defects in the ejaculate. Thus, globozoospermia is a severe reproductive issue requiring urgent resolution [3,4,5,6].
Previous studies have suggested that gene mutations might be the pathology underlying human globozoospermia, and several genes, such as DNAH6 [7] and SPATA16 [8], cause globozoospermia. DPY19L2 (DPY-19-like 2, MIM 613893) mutations affect sperm head elongation and acrosome formation and accounts for approximately 75% of the patients with globozoospermia [9,10,11,12,13]. SPATA16 was the first gene reported to be involved in the pathogenesis of globozoospermia in humans [8]. SPATA16 are located in Golgi body and anterior acrosome vesicles that are transported to the anterior segment to form acrosome during the formation of the spermatozoon, suggesting a key role of the protein in the formation of acrosome [8, 14]. SPATA16 mutations have been identified in some acrosome absent cases that result in globozoospermia as the membrane is unable to bind the zona pellucida and fertilize the oocyte [8, 14, 15]. PICK1 gene encoding a cytosolic protein is found in the proacrosomal vesicles of round spermatids; the mutations in this gene lead to the failure of the merge of proacrosomal vesicles, thereby resulting in globozoospermia [16,17,18]. CCIN encodes a major basic protein of the mammalian sperm head cytoskeleton, and its absence or altered arrangement of the calicin protein is related to globozoospermia [19, 20]. In mouse models, the knockout of Gopc [21], Hrb [22, 23], Csnk2a2 [24], Hsp90beta1 [25], Vps54 [26], Mfsd14a [27], and GM130 [28] can replicate the round-headed sperm phenotype, but whether these gene mutations also cause this phenotype in humans needs to be investigated further. Although the mutations of DPY19L2 account for up to 75% of globozoospermia, the genetic mutation spectrum is not yet completely fully elucidated, necessitating an in-depth investigation to deduce the involvement of DPY19L2 in globozoospermia.
In the present study, we investigated an infertile patient with globozoospermia and identified a homozygous deletion mutation in DPY19L2. This study confirmed that DPY19L2 mutation is the main cause of globozoospermia, which broadened the mutation spectrum of the gene.
Case presentation
Clinical presentation and family history
The proband (aged 27 years, II:1) and his family were recruited from the First Affiliated Hospital of Xiamen University. Pedigree analysis revealed recessive autosomal (AR) inheritance (Fig. 1a). He had sexual life 2–3 times/ week with normal erection and ejaculation during the last 4 years after marriage, but his wife did not conceive. The patient did not have any bad chemical contact history or habits such as smoking and drinking. The patient was in good health with no obvious discomfort. Physical examination data were as follows: height, 174 cm; weight, 66 kg; external genital development, normal; bilateral testicular size and bilateral spermatic vein, normal. The semen was light yellow and could be liquefied within 30 min. The examination results from our hospital were as follows: semen volume, 5.5–6.0 mL; semen pH, 7.2; sperm density, 9.1–11.3 × 106/mL; prorsad percentage motility, 15–18%; non-prorsad percentage motility, 12–15%. Sperm morphology examined by Papanicolaou staining showed 0% normal morphology. The biochemical testing of seminal plasma showed that the content of sperm acrosomase was 32.6 uIU/10 × 6; neutral glycosidase, 20.6 mU; fructose level, 17.5 μmol; zinc level, 5.4 μmol. The reproductive hormones were within normal ranges (follicle-stimulating hormone (FSH) 4.56 mIU/mL, luteinizing hormone (LH) 5.87 mIU/mL, T 4.34 ng/mL, E2 33 pg/mL, PRL 12.62 ng/mL). B-ultrasound showed no abnormalities in the seminal vesicles, prostate, bilateral testes, epididymis, and spermatic veins. The chromosomal karyotype of the patient was normal, 46, XY, and no microdeletions were detected in the Y chromosome. The patient’s family had yet to undergo follow-up treatment.

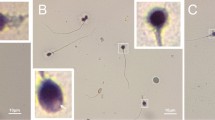
Consanguineous family analysis and phenotypes of patients with globozoospermia. a. Consanguineous pedigree of the proband family with complete deletion ofDPY19L2. The patient (II:1) was suffering from globozoospermia had homozygous deletion of exons 1, 11 and 22 of DPY19L2. And heterozygous deletion in exons 1, 11, 22 of DPY19L2 in mother of proband (I:2), while no mutation was found in exons 1, 11 and 22 of DPY19L2 of his father (I:1). b. Papanicolaou staining of sperm cells from normal control and the proband globozoospermia. Black arrows indicate spermatozoa from globozoospermia. Scale bar: 20 μm. c. Ultrastructure of the Sperm from normal control and patient with complete loss of DPY19L2 showing that the DPY19L2-null spermatozoa had round sperm head. The black arrow indicates the head of normal control sperm and the head of DPY19L2 completely missing sperm.
A volume of 5 mL peripheral blood was withdrawn from the patient, and each of his parents. The control subject was a healthy male, aged 28 years, with normal fertility. Written informed consent was obtained from each participant. This study was approved by the Ethics Committee of the First Affiliated Hospital of Xiamen University.
Mutations in DYP19L2 in the patient with globozoospermia
In order to determine the genetic variants associated with globozoospermia, we performed copy number variation sequencing (CNVseq) on the DNA samples of the proband and his parents. The mutation was detected in the DYP19L2 gene (Fig. 1). Approximately 180 kbp of homozygotic deletion was found on chromosome 12 locus 12q14.2 in the patient sample (g.63950001–64,130,000) (Fig. 2a). The functional gene in this segment included in the Decipher database was DPY19L2, which is related to spermatogenesis disorder. The main clinical phenotype is round head sperm, which eventually leads to male infertility.

Genome sequencing results of patient samples. a. The results of CNVseq: CNV: copy number variation. b. The domains and mutations inDPY19L2. The DPY19L2 is located on chromosome 12q14.2 and approximately 110.027 kb long. This gene has 22 exons encoding 758 amino acids with a molecular mass of 87.374 kDa. The Chromosome 12q14.2 (g.63950001–64130000) was homozygous deletion of about 180 kb,the mutation leads to complete deletion of DPY19L2
Additionally, the verification by CNV confirmed the homozygous deletion of exons 1, 11, and 22 of DPY19L2 in the proband. A similar phenotype was detected in the mother’s genome, while no such variation was found in the father’s genome (Supplementary Table 1).
Papanicolaou staining showed that the patient’s sperms had abnormal head development. As shown by the black arrow, the most common sperm defect in the patient was the appearance of the round head (Fig. 1b). Based on these results, the patient was diagnosed with globozoospermia (Fig. 1a, II:1).
Transmission electron microscopy (TEM) further confirmed these defects, and the sperm from the patient showed numerous ultrastructural defects in the head. The ultrastructure of the sperm in the control patient was normal, while that of the proband was round (Fig. 1c).
We determined the levels of DYPL92 in the sperm using Western blotting and found that DYP19L2 was not expressed in the sperm of the patient (Fig. 3).

Expression of DPY19L2 in the Patient and normal control. Western blotting analysis of DPY19L2 in sperm cells from control and the proband. Ac-tubulin was used as a loading control
Discussion and conclusion
Globozoospermia is a disease of sperm malformation characterized by round head sperm and lack of acrosome. These sperms cannot penetrate the zona pellucida of the oocyte, leading to failed fertilization and infertility [29]. When the sperms are injected into the cytoplasm, the fertilization rate tends to be low. Hitherto, the causes of the disorder remain unclear. DPY19L2 is a testis-specific transmembrane protein that is highly expressed in the testes. It anchors the acrosomal membrane to the nuclear membrane and is required for sperm head elongation and acrosome formation during spermatogenesis. However, mutations in this gene lead to instability and loss of acrosome [4, 5, 11, 30]. DPY19L2 is a pathogenic gene underlying spermatogenic failure, which is an AR inheritance disorder; the homozygous variation might lead to morbidity [31]. In the current study, the data of Papanicolaou staining and electron microscopy provided sufficient evidence of this phenotype in the patient’s sperm. To determine the genetic variation that leads to male infertility, genome-wide CNVseq was performed on the patient presenting globozoospermia. The results showed that DPY19L2 was absent.
Previous studies have shown that DPY19L2 mutation types exhibit variations in frequency distribution, which might be attributed to the limited number of patients recruited or analyzed in these studies. Also, the frequency distribution of DPY19L2 mutation types may be different in pure Chinese patients with this AR genetic disease due to the change in social and cultural factors such as blood relationships [4, 32].
Furthermore, reverse transcription-polymerase chain reaction (RT-PCR) detected the tiny variation of DPY19L2 in the sample through sequencing; however, the large fragment variation of the gene could not be determined. Thus, other quantitative PCR methods such as MLPA could be utilized to detect the large fragment variation of the gene [4]. Nonetheless, due to the high homology between DYP19L2 and its pseudogene, selecting a highly specific probe with a unique sequence matching the specific DPY19L2 nucleotide to overcome the interference of DPY19L2 pseudogene is essential [4, 32]. Moreover, any loss of heterozygotes shown in MLPA should be further confirmed by a long-range PCR [4].
Herein study, we found that 180-kbp homozygous deletions (g.63950001–64130000) at 12q14.2 on chromosome 12 in the sample of the patient subjected to CNVseq, including complete loss of DYP19L2. Complete gene loss leads to complete loss of DYP19L2, which might result in complete loss of DYP19L2 function; thus, the sperm of the patient presents globozoospermia. Therefore, this study broadens the mutation spectra of DYP19L2 mutations that cause globozoospermia.
In summary, our findings confirm that mutations in DYP19L2 are the major causes of globozoospermia in humans. The homozygosity deletion of DYP19L2 affects the normal development of sperm head, leading to the typical globozoospermia phenotype. Therefore, the current study provides researchers and clinicians with updated information about sperm with globozoospermia.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- TEM:
-
Transmission electron microscopy
- AR:
-
Autosomal recessive
- RT-PCR:
-
Reverse transcription-polymerase chain reaction
- MLPA:
-
Multiplex ligation-dependent probe amplification
- CNVseq:
-
Copy number variation sequencing
References
Krausz C. Male infertility: pathogenesis and clinical diagnosis. Best Pract Res Clin Endocrinol Metab. 2011;25(2):271–85.
O'Flynn O'Brien KL, Varghese AC, Agarwal A. The genetic causes of male factor infertility: a review. Fertil Steril. 2010;93(1):1–12.
Dam AH, Feenstra I, Westphal JR, Ramos L, van Golde RJ, Kremer JA. Globozoospermia revisited. Hum Reprod Update. 2007;13(1):63–75.
Coutton C, Zouari R, Abada F, Ben Khelifa M, Merdassi G, Triki C, Escalier D, Hesters L, Mitchell V, Levy R, et al. MLPA and sequence analysis of DPY19L2 reveals point mutations causing globozoospermia. Hum Reprod. 2012;27(8):2549–58.
Harbuz R, Zouari R, Pierre V, Ben Khelifa M, Kharouf M, Coutton C, Merdassi G, Abada F, Escoffier J, Nikas Y, et al. A recurrent deletion of DPY19L2 causes infertility in man by blocking sperm head elongation and acrosome formation. Am J Hum Genet. 2011;88(3):351–61.
De Braekeleer M, Nguyen MH, Morel F, Perrin A. Genetic aspects of monomorphic teratozoospermia: a review. J Assist Reprod Genet. 2015;32(4):615–23.
Li L, Sha YW, Xu X, Mei LB, Qiu PP, Ji ZY, Lin SB, Su ZY, Wang C, Yin C, et al. DNAH6 is a novel candidate gene associated with sperm head anomaly. Andrologia. 2018;50(4).
Dam AH, Koscinski I, Kremer JA, Moutou C, Jaeger AS, Oudakker AR, Tournaye H, Charlet N, Lagier-Tourenne C, van Bokhoven H, et al. Homozygous mutation in SPATA16 is associated with male infertility in human globozoospermia. Am J Hum Genet. 2007;81(4):813–20.
Koscinski I, Elinati E, Fossard C, Redin C, Muller J, Velez de la Calle J, Schmitt F, Ben Khelifa M, Ray PF, Kilani Z, et al. DPY19L2 deletion as a major cause of globozoospermia. Am J Hum Genet. 2011;88(3):344–50.
ElInati E, Kuentz P, Redin C, Jaber S, Vanden Meerschaut F, Makarian J, Koscinski I, Nasr-Esfahani MH, Demirol A, Gurgan T, et al. Globozoospermia is mainly due to DPY19L2 deletion via non-allelic homologous recombination involving two recombination hotspots. Hum Mol Genet. 2012;21(16):3695–702.
Zhu F, Gong F, Lin G, Lu G. DPY19L2 gene mutations are a major cause of globozoospermia: identification of three novel point mutations. Mol Hum Reprod. 2013;19(6):395–404.
Coutton C, Abada F, Karaouzene T, Sanlaville D, Satre V, Lunardi J, Jouk PS, Arnoult C, Thierry-Mieg N, Ray PF. Fine Characterisation of a Recombination Hotspot at the DPY19L2 Locus and Resolution of the Paradoxical Excess of Duplications over Deletions in the General Population. PLoS Genet. 2013;9(3).
Ghazavi F, Peymani M, Hashemi MS, Ghaedi K, Nasr-Esfahani MH. Embryos derived from couples with consanguineous marriages with globozoospermia should be screened for gender or DPY19L2 deletion. Andrologia. 2019;51(4).
ElInati E, Fossard C, Okutman O, Ghedir H, Ibala-Romdhane S, Ray PF, Saad A, Hennebicq S, Viville S. A new mutation identified in SPATA16 in two globozoospermic patients. J Assist Reprod Genet. 2016;33(6):815–20.
Fujihara Y, Oji A, Larasati T, Kojima-Kita K, Ikawa M. Human Globozoospermia-Related Gene Spata16 Is Required for Sperm Formation Revealed by CRISPR/Cas9-Mediated Mouse Models. Int J Mol Sci. 2017;18(10).
Xiao N, Kam C, Shen C, Jin WY, Wang JQ, Lee KM, Jiang LW, Xia J. PICK1 deficiency causes male infertility in mice by disrupting acrosome formation. J Clin Invest. 2009;119(4):802–12.
He J, Xia MY, Tsang WH, Chow KL, Xia J. ICA1L forms BAR-domain complexes with PICK1 and is crucial for acrosome formation in spermiogenesis. J Cell Sci. 2015;128(20):3822–36.
Li YH, Zhang N, Wang YN, Shen Y, Wang Y. Multiple faces of protein interacting with C kinase 1 (PICK1): structure, function, and diseases. Neurochem Int. 2016;98:115–21.
Hess H, Heid H, Franke WW. Molecular characterization of mammalian Cylicin, a basic-protein of the sperm head cytoskeleton. J Cell Biol. 1993;122(5):1043–52.
Vonbulow M, Heid H, Hess H, Franke WW. Molecular nature of Calicin, a major basic-protein of the mammalian sperm head cytoskeleton. Exp Cell Res. 1995;219(2):407–13.
Bizkarguenaga M, Gomez-Santos L, Madrid JF, Saez FJ, Alonso E. Increase of germ cell nuclear factor expression in globozoospermic Gopc(−/−) knockout mice. Andrology. 2019;7(3):319–28.
Kang-Decker N, Mantchev GT, Juneja SC, McNiven MA, van Deursen JM. Lack of acrosome formation in Hrb-deficient mice. Science. 2001;294(5546):1531–3.
Juneja SC, van Deursen JM. A mouse model of familial oligoasthenoteratozoospermia. Hum Reprod. 2005;20(4):881–93.
Xu X, Toselli PA, Russell LD, Seldin DC. Globozoospermia in mice lacking the casein kinase II alpha' catalytic subunit. Nat Genet. 1999;23(1):118–21.
Audouard C, Christians E. Hsp90beta1 knockout targeted to male germline: a mouse model for globozoospermia. Fertil Steril. 2011;95(4):1475–7 e1471–1474.
Paiardi C, Pasini ME, Gioria M, Berruti G. Failure of acrosome formation and globozoospermia in the wobbler mouse, a Vps54 spontaneous recessive mutant. Spermatogenesis. 2011;1(1):52–62.
Doran J, Walters C, Kyle V, Wooding P, Hammett-Burke R, Colledge WH. Mfsd14a (Hiat1) gene disruption causes globozoospermia and infertility in male mice. Reproduction. 2016;152(1):91–9.
Han F, Liu C, Zhang L, Chen M, Zhou Y, Qin Y, Wang Y, Chen M, Duo S, Cui X, et al. Globozoospermia and lack of acrosome formation in GM130-deficient mice. Cell Death Dis. 2017;8(1):e2532.
Vicari E, Perdichizzi A, De Palma A, Burrello N, D'Agata R, Calogero AE. Globozoospermia is associated with chromatin structure abnormalities: case report. Hum Reprod. 2002;17(8):2128–33.
Pierre V, Martinez G, Coutton C, Delaroche J, Yassine S, Novella C, Pernet-Gallay K, Hennebicq S, Ray PF, Arnoult C. Absence of Dpy19l2, a new inner nuclear membrane protein, causes globozoospermia in mice by preventing the anchoring of the acrosome to the nucleus. Development. 2012;139(16):2955–65.
Noveski P, Madjunkova S, Maleva I, Sotiroska V, Petanovski Z, Plaseska-Karanfilska D. A homozygous deletion of the Dpy19l2 gene is a cause of Globozoospermia in men from the republic of Macedonia. Balkan J Med Genet. 2013;16(1):73–6.
Carson AR, Cheung J, Scherer SW. Duplication and relocation of the functional DPY19L2 gene within low copy repeats. BMC Genomics. 2006;7:45.
Acknowledgments
The authors greatly appreciate the participation of the family in this study.
Declarations
The authors declare no competing or financial interests.
Funding
This work was supported by the Natural Science Foundation of Fujian Province of China (Grant No. 2019J01565 and 2019J01566) and by the National Science Foundation of China (No. 81701419). Fujian Provincial Natural Science Foundation of China (approval No. 2019J01565 and 2019J01566) was mainly responsible for supporting the purchase of reagents, such as antibodies and related consumables. National Science Foundation of China (81701419) provided funds for experiments and data analysis.
Author information
Authors and Affiliations
Contributions
YZ recruited the family, coordinated the research, and performed genetic analyses. RW and XZ performed molecular experiments and drafted the manuscript. WL and YY performed molecular experiments. ZL and NL designed the study and performed data analyses. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The patient provided written consent before participation in the study. The Ethics Committee at the First Affiliated Hospital of Xiamen University approved the data for publication.
Consent for publication
The patient provided written consent for publication of their data.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplementary Table 1
. Results of exon mutation test on proband and his parents
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Yz., Wu, Rf., Zhu, Xs. et al. Identification of a novel deletion mutation in DPY19L2 from an infertile patient with globozoospermia: a case report. Mol Cytogenet 13, 24 (2020). https://doi.org/10.1186/s13039-020-00495-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-020-00495-1