
Abstract
Background
Few patients with interstitial deletions in the distal long arm of chromosome 14 have been reported, and these patients showed rather indistinct features, including growth and mental retardation and phenotypic alterations.
Results
We describe a de novo 14q interstitial deletion in a 6-year-old boy with dysmorphic facial traits such as hypertelorism, short and narrow palpebral fissures, broad nose with anteverted nostrils, long philtrum, thin upper lip with cupid’s bow, prominent and everted lower lip, mildly low-set ears, as well as moderate developmental delay and mild mental retardation. Array-CGH mapped the deletion to the region 14q24.3 to 14q31.3, including 13.11 Mb, proximal to the imprinted genomic region of 14q32.
Conclusion
This mild phenotypic presentation suggests that the deleted segment does not contain essential genes for early organ development. Twenty-two genes with known functions, including Neurexin III (NRXN3, OMIM 600567), map to the region deleted in the propositus.
Similar content being viewed by others
Background
The few reported patients with interstitial microdeletions in the long arm of chromosome 14 have shown a broad and inconsistent spectrum of clinical features caused by loss of different segments of 14q. An understanding of the phenotypes of these clinical disorders is further hampered by the scarcity of reports and the fact that most of the reported patients were diagnosed using only traditional chromosome banding with inaccurate and approximate mapping of the deleted segment.
Cingöz et al. [1], considering the minimal size of these deletions in 11 patients, constructed a map for the three shortest regions of deletion overlap. The proximal region includes the gene encoding Neurexin III (NRXN3, MIM 600567), for which haploinsufficiency is associated with cognitive and neurological defects.
Here, we report a male patient with a de novo deletion of the segment 14q24.3 to q31.3, determined using comparative genomic hybridization with microarray (array CGH; aCGH). We also sought to correlate the clinical findings to those of patients with deletions overlapping that identified in the propositus. Considering that breakpoint determination using only banded karyotypes is not precise and may be inaccurate, we included only reports with molecular determination or confirmation of the breakpoints in our comparison.
Case presentation
The patient was a male aged 6 years and 9 months, the only son of a 38-year-old mother and a 41-year-old father who measured 1.58 m and 1.76 m in height, respectively, and were healthy and nonconsanguineous. The parents reported no genetic conditions or birth defects in the family. The pregnancy was planned and uneventful. Delivery was by cesarean section at 39 weeks and 6 days. The patient’s birth weight was 3145 g, with a length of 49 cm and head circumference of 33.5 cm, which are all appropriate for the gestational age. There was no visible perinatal asphyxia. The postnatal course was characterized by psychomotor delay and muscular hypotonia.
At the first clinical evaluation at 20 months of age, he showed mild dysmorphic features. The first tooth erupted at 8 months of age. Beginning at 6 months of age, he received regular neurological and pediatric follow-ups. The first smile was noted at the age of 2 months, rolling from a ventral to a dorsal position was observed at 3 months, following objects with his eyes was observed at 6 months, maintaining a sitting position was observed at 8 months, maintaining a standing position was observed at 10 months, responding to his name was observed at 10 months, walking was observed at 20 months, and toilet training was achieved at 2 ½ years.
At 6 months of age, physical therapy and an interdisciplinary program of early stimulation were initiated. He was discharged from physical therapy at the end of the second year of life. Psychologic evaluation at 2 years of age showed cognitive delay (corresponding to 1 year and 6 months). He was subjected to speech therapy due to difficulties related to sensory-motor aspects and oral language. According to a clinical assessment at 4 years of age, despite having communicative intent, language expression was quite restricted. He also had deficits in psychomotor and cognitive executive function that improved with time, especially regarding sensory integration, and he presented an attention deficit. At 5 years of age, an evaluation of language and cognitive development was performed using the Protocol of Observation Behavioral [2]. The examinations revealed the following: limited oral language, with attempts to engage in dialogue; language disorder, especially in expressive language; deficit in motor skills and difficulties in the understanding and execution of tasks and games. With respect to activities of daily living, he showed autonomy in eating and dressing.
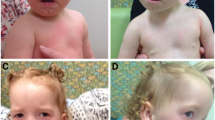
At 6 years and 10 months of age, he measured 125 cm in height (50th – 75th percentile), weighed 26 kg (50-75th percentile) and had an occipital frontal circumference (OFC) of 52 cm (25th – 50th percentile). Dysmorphological examination revealed low-set ears with prominent helices and lobules, hypertelorism (ICD 3.5 cm, >97th percentile), bushy eyebrows, short nose with anteverted nostrils, deep nasolabial furrows, small and open mouth with an open bite and a thin vermilion, a prominent Cupid’s bow of the upper lip and a prominent and everted lower lip. Mild micrognathia was also observed (Figure 1).

Patient at the ages of 2 months (A), 3 years and 5 months (B) and 6 years and 10 months (C). Note the short upturned nose, deep philtrum, thin vermillion of the upper lip and prominent earlobes.
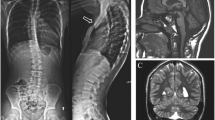
The propositus attends a regular school with support. He communicates well with colleagues and teachers, and he is able to express his wishes through language and to understand simple conversation and demands. Further examinations revealed the following: Magnetic resonance imaging (MRI) showed an intact corpus callosum and a small region of ischemia in the glyptic cavity of the cerebellar region. Electroencephalography (EEG) and audiometry results were normal, and he showed no vision problems.
Results
With regard to cytogenetic and molecular examinations, the deletion was initially identified through G-banding. The karyotype was interpreted as 46,XY,del(14)(q31). However, fluorescence in situ hybridization (FISH) performed in the proband showed that there was no loss of the terminal region of chromosome 14 (band 14q32). The precise characterization of the deletion using oligonucleotide aCGH showed a loss of genomic material corresponding to an interstitial deletion in the long arm of chromosome 14, segment q24.3-q31.3, of approximately 13.11 Mb (UCSC Genome Browser on Human Feb. 2009 (GRCH37/hg19). Arr 14q24.3q31.3 (76,211,806-89,327,656) ×1 (Figure 2). The parental karyotypes were normal.

Chromosome 14 aCGH profile of the patient (in red) and a reference DNA sample from a normal male (in blue). The figure shows (A) a copy number loss corresponding to the segment 14q24.3-q31.3 in a genomic segment with the median log2 ratio shifted to -0.8 (red line). The probe log2 ratios were plotted according to genomic coordinates (based on the UCSC Genome Browser, February 2009, NCBI Build 37 reference sequence). In (B), detail of the 14q24.3-q31.3 region showing the deleted region of ~13 Mb.
Discussion
Excluding pseudogenes, chromosome 14 presents a mean gene density of 10 genes per Mb (850 genes per 85 Mb), which is close to the mean value estimated for the whole human genome (9.3–10.8 genes per Mb) [3]. Mutation analysis has revealed 21 genes on this chromosome associated with genetic diseases [4]. Among these, Holt-Oram syndrome or a syndrome similar to Holt-Oram syndrome, has been reported in patients with 14q deletions. Furthermore, 70% of those patients fulfilling strict criteria show a mutation in TBX5 on 12q24.1. Thus, there might be a second locus for this syndrome at 14q23.3q31.1 [5].
The propositus presented with mild to moderate developmental delay, normal growth and a pattern of indistinct minor dysmorphic signs in the absence of organ malformations. For a comparison with previously reported patients with similar or overlapping deletions spanning at least 10 Mb (see Table 1, in order from proximal to distal from the proximal breakpoint), we excluded reports in which the breakpoints were determined only through chromosome banding because it is well known that such methods can provide imprecise determinations leading to incorrect interpretations (for example, a deletion classified as terminal may turn out to be interstitial, as in our proband, and vice versa). Our patient was the first to be subjected to aCGH analysis of the deleted segment. In contrast, using FISH with only a few and mostly flanking markers, it is possible to overlook non-deleted or even duplicated stretches of DNA within a deleted segment, especially in instances of ring chromosomes [6].
Table 1 shows that except for the patient described by LeMeur [5], the proximal breakpoints were quite similar in all patients, ranging from bp 73.942.355 [7], patient 25 to 77,867,749 [7], patient 22, whereas the distal breakpoints differed considerably, from 88,488,190 [8],[9] to 98.771.224 [1],[10]. However, these represent the minimal deleted segments because the space between the last or first present marker and the first or last deleted marker was quite different, and some markers were not precisely mapped. In addition, findings present in only one patient were listed separately, and comparison was limited by the uncertainty of whether dysmorphic findings not described in the different case reports were absent or not considered worth mentioning.
Our comparison of clinical features revealed that neurodevelopmental features such as developmental delay, muscular hypotonia and language delay were quite consistent, as were some frequent and non-specific dysmorphic signs including hypertelorism, inner epicanthic folds, short and bulbous nose with a depressed bridge, thin upper lip and dysplastic ears. However, malformations were inconsistently found, as heart defects were only found in 3/6 patients, and different defects were observed in one patient each. Hearing impairment was reported 4 times, but the other studies did not report on the presence or absence of hearing impairment, and in the reported cases, the type was conductive and sensory-neural, with 2 instances each.
The patient with the greatest accumulation of congenital malformations had at least 6 Mb deleted proximal of the deletion in the other patients [5]. We did not include the following two patients in the comparison. First, Vaags et al. [11] reported a patient with a very small deletion (0.063 Mb) inside that detected in our propositus; this patient had Asperger syndrome with NRXN3 deletion. Although NRXN3 is associated with autism [12] and lies inside our reported deletion, our propositus did not display features of autism. Second, Shimada et al. [12] described a 2-year-old boy with growth and severe mental retardation, a dysmorphic pattern comparable to that observed in our patient and MRI findings of leukodystrophy. This deletion spanned 2.0 Mb, of which 0.8 Mb overlapped with the proximal deleted region in our patient. This patient was found to have a deletion of EIF2B2, a gene associated with leukodystrophy, which maps outside the deletion in our propositus, who consequently does not show similar neurologic symptoms.
Table 2 lists the genes with only one copy in the propositus. The list shows that mutations of some of these genes, including TTC8 (Bardet-Biedl syndrome 8), POMT2 (Walker-Warburg syndrome), GALC (Krabbe disease), GSTS1 (Tyrosinemia Ib), and SPATA7 (Leber amaurosis 3), produce well-known clinical entities. However, findings characteristic for these conditions are not present in our propositus because hemizygosity would not cause the condition unless the remaining allele carries a mutation or in the case of uniparental disomy. Except for NRXN3 and EIF2B2 (see above), the genes listed in Table 2 are not known to cause specific phenotypes if one allele is absent. This is different from more proximal deletions that cause anophthalmia, cataract (SIX1, BMP4)), hearing loss (OTX2), abnormal pituitary function (SIX4 and SIX6) and polydactyly, all due to hemizygosity of the related genes [13]-[16]. In patients with deletions distal from that in the propositus and mostly terminal, including ring chromosomes, the clinical patterns tend to be indistinct, mostly showing patterns of dysmorphic signs [17],[18]. One exception is the case of a mother and daughter who both showed hemifacial microsomia [19]; however, the latter could theoretically be independent of the small deletion.
Genomic imprinting could theoretically cause part of the clinical picture of our propositus. Sutton and Schaffer [20] suggested that 14q23-q32 is likely an area where imprinted genes may reside because distinct phenotypes have been associated with both maternal and paternal uniparental disomy (UPD) for this chromosome. However, according to the authors, these features mapped to other segments, and they were not present in the propositus. Unfortunately, we could not determine the parental origin of the deletion in our propositus. Generally, most interstitial deletions are paternal in origin. However, because maternal UPD is correlated with profound intrauterine and postnatal growth deficiency and early puberty, loss of a critical imprinted segment in the region 14q24-q31 is unlikely because the propositus does not show this key feature. In addition, the clinical picture of paternal UPD 14 is more severe and less consistent; therefore, a comparison with our patient was not possible.
Conclusions
In summary, the clinical findings in our propositus compared to those in patients with similar deletions show that deletion of the segment 14q24.3-q31.3 is accompanied by a pattern of developmental delay, muscular hypotonia and a relatively non-specific pattern of dysmorphic findings. Thus, it would be difficult or even impossible to recognize similar cases on the basis of clinical findings alone.
Methods
Standard GTG banding was performed at a resolution of 400-550 bands on metaphase chromosomes from peripheral blood lymphocytes of the patient and his parents. Molecular karyotyping was performed on the proband using through whole-genome array-CGH using a 60-mer oligonucleotide-based microarray with a theoretical resolution of 40 kb (8x60K, Agilent Technologies Inc., Santa Clara, CA) (Figure 2A,B). Labeling and hybridization were performed following the protocols provided by the manufacturers. Images of the arrays were taken using a microarray scanner (G2600D) and processed using Feature Extraction software (v9.5.1), both from Agilent. The raw data were analyzed by Agilent Cytogenomics v2.7.8.0 software with the statistical algorithm ADM-2, using a threshold of 6.0 and a 4-probe minimum aberration call.
Consent
Written informed consent was obtained from the patient’s parents for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
References
Cingöz S, Blache I, Bjerglund L, Ropers H-H, Tommerup N, Jensen H, Brondum-Nielsen K, Tümer Z: Interstitial deletion of 14q24.3-q32.2 in a male patient with plagiocephaly, BPES features, developmental delay, and congenital heart defects. Am J Med Genet Part A 2011, 155: 203–206. 10.1002/ajmg.a.33766
Zorzi JL, Hage SRV: PROC- Protocolo de observação comportamental: avaliação de linguagem e aspectos cognitivos infantis. Pulso Editorial, São José dos Campos; 2004.
Heilig R, Eckenberg R, Petit JL, Fonknechten N, Da Silva C, Cattolico L, Levy M, Barbe V, de Berardinis V, Ureta-Vidal A, Pelletier E, Vico V, Anthouard V, Rowen L, Madan A, Qin S, Sun H, Du H, Pepin K, Artiguenave F, Robert C, Cruaud C, Brüls T, Jaillon O, Friedlander L, Samson G, Brottier P, Cure S, Ségurens B, Anière F, et al.: The DNA sequence and analysis of human chromosome 14. Nature 2003,421(6923):601–7. 10.1038/nature01348
Kamnasaran D, Cox DW: Current status of human chromosome 14. J Med Genet 2002, 39: 81–90. 10.1136/jmg.39.2.81
Le Meur N, Goldenberg A, Michel-Adde C, Drouin-Garraud V, Blaysat G, Marret S, Amara SA, Moirot H, Joly-Hélas G, Mace B, Kleinfinger P, Saugier-Veber P, Frébourg T, Rossi A: Molecular characterization of a 14q deletion in a boy with features of Holt-Oram syndrome. Am J Med Genet A 2005,2005(134):439–442. 10.1002/ajmg.a.30660
Rossi E, Riegel M, Messa J, Gimelli S, Maraschio P, Ciccone R, Stroppi M, Riva P, Perrotta C, Mattina T, Baumer A, Kucinskas V, Castellan C, Schinzel A, Zuffardi O: Duplications in addition to terminal deletions are present in a proportion of ring chromosomes: clues to the mechanisms of formation. J Med Genet 2008, 45: 147–54. 10.1136/jmg.2007.054007
Zollino M, Seminara L, Orteschi D, Gobbi G, Giovannini S, Della Giustina E, Frattini D, Scarano A, Neri G: The ring 14 syndrome: clinical and molecular definition. Am J Med Genet A 2009, 149: 1116–1124. 10.1002/ajmg.a.32831
Karnitis SA, Burns K, Sudduth KW, Golden WL, Wilson WG: Deletion (14) (q24.3q32.1): evidence for a distint clinical phenotype. Am J Med Genet 1992, 44: 153–157. 10.1002/ajmg.1320440207
Byth BC, Costa MT, Teshima IE: Molecular analysis of three patients with interstitial deletions of chromosome band 14q31. J Med Genet 1995, 32: 564–567. 10.1136/jmg.32.7.564
Schlade-Bartusiak K, Macintyre G, Zunich J, Cox DW: A child with deletion (14)(q24.3q32.13) and auditory neuropathy. Am J Med Genet A 2008, 146: 117–123. 10.1002/ajmg.a.32064
Vaags AK, Lionel AC, Sato D, Goodenberger MK, Stein QP, Curran S, Ogilvie C, Ahn JW, Drmio I, Senman L, Chrysler C, Thomoson A, Russel C, Prasad A, Walker S, Pinto D, Marshall C, Stravopoulos D, Zwaigenbaum L, Fernandez BA, Fombonne E, Bolton P, Collier DA, Hodge JC, Roberts W, Szatman P, Scherer SW: Rare deletions at the Neurexin 3 locus in Autism Spectrum Disorder. Am J Hum Genet 2012, 90: 133–141. 10.1016/j.ajhg.2011.11.025
Shimada S, Miya K, Oda N, Watanabe Y, Kumada T, Sugawara M, Shimojima K, Yamamoto T: An unmasked Mutation of EIF2B2 due to submicroscopic deletion of 14q24.3 in a patient with vanishing white matter disease. Am J Med Genet A 2012, 158: 1771–1777. 10.1002/ajmg.a.35431
Guilherme RS, Cernach MCSP, Sfakianakis TE, Takeno SS, Nardizza LMM, Rossi C, Bhatt SS, Liehr T, Melaragno MI: A complex chromosome rearrangement involving four chromosomes, nine breakpoints and a cryptic 0.6-Mb deletion in a boy with cerebellar hypoplasia and defects in skull ossification. Cytogenet Genome Res 2013, 141: 317–323. 10.1159/000353302
Oehl-Jaschkowitz B, Vanakker OM, De Paepe A, Menten B, Martin T, Weber G, Christmann A, Krier R, Scheid S, McNerlan SE, McKee S, Tzschach A: Deletions in 14q24.1q24.3 are associated with congenital heart defects, brachydactyly, and mild intellectual disability. Am J Med Genet A 2014, 164: 620–626. 10.1002/ajmg.a.36321
Brisset S, Slamova Z, Dusatkova P, Briand-Suleau A, Milcent K, Metay C, Simandlova M, Sumnik Z, Tosca L, Goossens M, Labrune P, Zemankova E, Lebl J, Tachdjian G, Sedlacek Z: Anophthalmia, hearing loss, abnormal pituitary development and response to growth hormone therapy in three children with microdeletions of 14q22q23. Mol Cytogenet 2014, 7: 17–23. 10.1186/1755-8166-7-17
Martinez-Frias ML, Ocejo-Vinyals JG, Arteaga R, Martinez-Fernandez ML, MacDonald A, Pérez-Belmonte E, Bermejo-Sanchez E, Martinez S: Interstitial deletion 14q22.3q23.2: genotype-phenotype correlation. Am J Med Genet A 2014, 164: 639–647. 10.1002/ajmg.a.36330
Yen FS, Podruch PE, Weisskopf B: A terminal deletion (14)(q31.1) in a child with microcephaly, narrow palate, gingival hypertrophy, protuberant ears and mild mental retardation. J Med Genet 1989, 26: 130–133. 10.1136/jmg.26.2.130
Piccione M, Antona V, Scavone V, Nalacarne M, Pierluigi M, Grasso M, Corsello G: Array CGH defined interstitial deletion on chromosome 14: a new case. Eur J Pediatr 2010, 169: 845–851. 10.1007/s00431-009-1128-4
Gimelli S, Cuoco C, Ronchetto P, Gimelli G, Tassano E: Interstitial deletion 14q31.1q31.3 transmitted from a mother to her daughter, both with features of hemifacial microsomia. Hum Genet 2013, 54: 361–364.
Sutton VR, Schaffer LG: Search for imprinted regions on chromosome 14: comparison of maternal and paternal UPD cases with cases of chromosome 14 deletion. Am J Med Genet 2000,2000(93):381–387. 10.1002/1096-8628(20000828)93:5<381::AID-AJMG7>3.0.CO;2-9
Acknowledgments
The authors thank the patient's family for their kind cooperation in all stages of this study. The investigation was supported by the Brazilian Network of Reference and Information in Microdeletion Syndromes (RedeBRIM), CNPq/Brazil grant number: 402012/2010-0 and Swiss National Foundation, Grant number: 320030-113635.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors have made substantial contributions to conception and design, acquisition of data, analysis and interpretation of data. All authors have been involved in drafting the manuscript and revising it critically for important intellectual content. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
Cite this article
Riegel, M., Moreira, L.M., Espirito Santo, L.D. et al. Interstitial 14q24.3 to q31.3 deletion in a 6-year-old boy with a non-specific dysmorphic phenotype. Mol Cytogenet 7, 77 (2014). https://doi.org/10.1186/s13039-014-0077-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-014-0077-4