
Abstract
Ever since a GGGGCC hexanucleotide repeat expansion mutation in C9ORF72 was identified as the most common cause of familial amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), three competing but nonexclusive hypotheses to explain how this mutation causes diseases have been proposed and are still under debate. Recent studies in the field have tried to understand how the repeat expansion disrupts cellular physiology, which has suggested interesting convergence of these hypotheses on downstream, functional defects in cells, such as nucleocytoplasmic transport disruption, membrane-less organelle defects, and DNA damage. These studies have not only provided an integrated view of the disease mechanism but also revealed novel cell biology implicated in neurodegeneration. Furthermore, some of the discoveries have given rise to new ideas for therapeutic development. Here, we review the research progress on cellular pathophysiology of C9ORF72-mediated ALS and FTD and its therapeutic implication. We suggest that the repeat expansion drives pathogenesis through a combination of downstream defects, of which some can be therapeutic targets.
Similar content being viewed by others
Background
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are two fatal neurodegenerative diseases with no curative treatment. ALS is a motor neuron degenerative disease with a lifetime risk of one in 347 men and one in 436 women, whereas FTD is the most common form of dementia for people under the age of 45 [1, 2]. ALS is characterized by degeneration of upper and lower motor neurons, causing muscle weakness and atrophy, whereas FTD is characterized by frontotemporal lobar degeneration, which can cause a heterogeneous group of clinical presentations, including progressive declines in cognition, behavior, and/or language. Based on the clinical presentations, FTD can be categorized into three subtypes: behavioral variant FTD, semantic dementia, and progressive non-fluent aphasia [3, 4].
Both ALS and FTD are associated with cellular deposition of protein inclusions. For ALS, a pathological hallmark observed in ~98% cases is the mislocalization and aggregation of TAR-DNA binding protein 43 (TDP-43). Other less frequently observed neuropathology includes the aggregation of superoxide dismutase 1 (SOD1), fused in sarcoma (FUS), and some heterogeneous nuclear ribonucleoproteins (hnRNPs) ([5,6,7,8];). For FTD, ~40% cases exhibit TDP-43 pathology, and another ~40% cases exhibit pathology of microtubule-associated protein tau. Other less frequently observed neuropathology includes FUS pathology and ubiquitin-positive aggregates with the protein components uncharacterized [8,9,10].
Despite their symptomatic differences, ALS and FTD share clinical, neuropathological, and genetic features and are part of a common spectrum. Indeed, ALS and FTD can occur in the same family, and many patients develop signs of both diseases. Furthermore, both ALS and FTD can be related to TDP-43 and FUS neuropathology [9]. In addition, mutations in several genes, including chromosome 9 open reading frame 72 (C9ORF72), have been identified to cause both ALS and FTD [11,12,13,14,15].
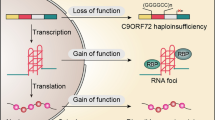
A GGGGCC (G4C2) hexanucleotide repeat expansion (HRE) in C9ORF72 is the most common genetic cause of familial ALS (40%) and FTD (25%) and also presents in some sporadic cases (ALS: 8%; FTD:5%). The lengths of G4C2 HREs are greater than 30 in most patients but vary among individuals, with some patients carrying >1,000 repeats [12, 14]. How the G4C2 HRE causes neurodegeneration is not fully understood. Past studies have suggested that the toxicity arises from one or more of the following assaults (Figure 1A): 1) loss of C9ORF72 due to aborted transcription, 2) bi-directionally transcribed G4C2 and G2C4 repeat RNAs from the HREs [16, 17], and/or 3) dipeptide repeat proteins (DPRs) translated from the repeat RNAs, via repeat-associated, non-ATG (RAN) translation [18,19,20,21,22]. As the DPR translation is ATG-independent, it occurs in all three frames bi-directionally, leading to five different DPR species: poly-(glycine-alanine, or GA) and (glycine-arginine, or GR) from the sense (G4C2) transcript, poly-(proline-alanine, or PA) and (proline-arginine, or PR) from the antisense (G2C4) transcript, and poly-(glycine-proline, or GP) from both the sense and antisense transcripts.

Summary of current cellular pathophysiological studies on C9ALS/FTD. a Three hypothesized primary assaults caused by the C9ORF72 mutation: 1) loss of C9ORF72 function, 2) repeat RNA forming either G-quartets or R-loops, toxic secondary structures that either sequester RBPs or cause DNA damage, respectively, and 3) DPRs. b The three primary assaults cause downstream, functional defects in nerve cells, and a combination of these defects causes neurodegeneration. c Therapeutic approaches can target either the primary assaults themselves, or their downstream effectors.
Consistent with this idea, loss of C9ORF72 mRNA and proteins, G4C2, G2C4 repeat RNA foci, and aggregation of DPRs have been observed in patient tissues and model systems. Furthermore, some of these assaults can indeed cause neurodegeneration and/or are cytotoxic in certain model systems. However, other studies also suggest evidence against any of these three hypotheses. These studies, with a goal of resolving the debate on these three assaults, have been extensively reviewed by others [23,24,25,26,27].
Besides research efforts to resolve this debate, recent studies on C9ORF72-mediated ALS/FTD (C9ALS/FTD) have related the three assaults to downstream, functional defects in cells. These studies identified molecular and cellular events that are crucial for neurodegeneration implicated in C9ALS/FTD, suggesting novel therapeutic targets for the disease (Figure 1 B and C). Moreover, these findings have greatly improved our understanding of fundamental cell biology beyond the scope of neurodegenerative diseases. Here, we review recent studies on C9ALS/FTD from a cell biology perspective, with a focus on cellular pathophysiology and therapeutic implications.
Main Text
Model Systems to Study C9ALS/FTD Cellular Pathophysiology
To study the pathomechanism of C9ALS/FTD, many cellular and animal models have been used, including yeast, C. elegans, Drosophila, zebrafish, mouse, and neurons derived from patient induced pluripotent stem (iPS) cells (iPSNs). Using these model systems, research has identified critical cellular events in C9ALS/FTD pathogenesis. We will review the strengths and contributions of different model systems in C9ALS/FTD research.
Yeast and Drosophila
Yeast and Drosophila do not have a C9ORF72 homolog. However, their short generation time and ease to handle make them powerful genetic tools to study the gain-of-toxicity mechanism. Many yeast or fly models of C9ALS/FTD have been established by ectopically expressing the G4C2 repeat RNA and/or DPRs, which causes cell death or neurodegeneration [12, 28,29,30,31,32,33,34,35]. Studies in these models have related the C9ORF72 gain of toxicity to arginine-containing DPRs [29, 33, 34]. Furthermore, large-scale genetic screens in these models have identified crucial pathogenic events [28, 29, 32, 36, 37] and proteins involved in the production of the repeat RNAs or DPRs [30, 31, 38,39,40]. Importantly, these findings have been further verified in higher model organisms and patients, suggesting the power of yeast and Drosophila in studying the C9ALS/FTD disease mechanism.
Mouse
Mouse C9orf72 is homologous to human C9ORF72 and thus, its knockout (KO) can be used to study the loss-of-function mechanism. However, mouse C9orf72 does not contain G4C2 repeats. Thus, one must ectopically express the repeat RNAs or DPRs in mice, as in yeast and Drosophila, to study the gain-of-toxicity mechanism. Several loss- or gain-of-function mouse models have been established for C9ALS/FTD [41,42,43,44,45,46], among which the models with adeno-associated-virus (AAV)-mediated repeat RNA or DPR expression have shown robust motor neuron loss and locomotion defects. Studies in these models have not only validated findings from yeast, Drosophila, and cultured cells [47,48,49,50] but also discovered novel disease pathophysiology [51].
IPSN
So far, translating drugs identified in animal models to ALS/FTD patients have gained little success [52,53,54], suggesting a gap between animal research and clinical studies. The recently developed iPS technology provides a potential solution to fill this gap [55, 56], as the iPSNs are derived from patients. Using iPSNs as models, studies on C9ALS/FTD have not only validated findings from animal models but also identified important pathophysiological events, such as ER stress [57]. Furthermore, these iPSNs provide a platform for genetic and drug screens [58, 59], which can be done in the future.
Other Model Systems
Besides yeast, fly, mouse and iPSN models, C. elegans and zebrafish models have also been established to study the C9ALS/FTD mechanism [60,61,62,63,64,65]. These studies have provided insights into both the loss- and gain-of-function mechanisms.
Using Multiple Model Systems
A major challenge in disease research is that all model systems have limitations. Thus, validation across model systems has been a powerful approach in studying human disease pathogenesis. Since non-vertebrate models are quick and easy to handle, whereas mouse and iPSN models are more disease-relevant, an efficient strategy to study disease mechanism is to first use non-vertebrate models to identify potential mechanisms and then, validate the findings in mammals and patient-derived iPSNs. This strategy ensures both the quickness and disease relevance and has been very successful in studying the C9ALS/FTD pathophysiology.
C9ALS/FTD Mechanisms
Loss of function
Consistent with the loss-of-function hypothesis, C9ORF72 transcript levels are decreased in some C9ALS/FTD patients [12, 66,67,68,69,70]. However, whether and how C9ORF72 loss contributes to pathogenesis is unclear. While C9ORF72 knockdown causes motor neuron degeneration in C. elegans and zebrafish [60, 65], C9ORF72 KO mice do not exhibit neurodegeneration or motor deficits [42, 45, 71,72,73], suggesting that C9ORF72 loss is insufficient to cause the disease in mammals. However, this does not exclude a contribution of C9ORF72 loss to pathogenesis. Indeed, recent studies in patient iPSNs or mice suggested that C9ORF72 loss can synergize with its gain of toxicity—C9ORF72 KO enhances the sensitivity of iPSNs to toxic DPRs [74], and C9ORF72 loss exacerbates neurodegeneration caused by HRE gain of toxicity in mouse models [75]. The latter study also shows that C9ORF72 loss disrupts autophagy, accompanied by an increase in DPR levels [75], raising the possibility that C9ORF72 loss compromises the autophagolysosomal clearance of DPRs.
Cellular Vesicle Trafficking Impairment
C9ORF72 protein is structurally similar to Rab guanine nucleotide exchange factors (RabGEFs) and functions as a RanGEF when complexing with two other proteins, WDR41 and SMCR8 [76]. RabGEFs activate Rab proteins, a group of small GTPases that regulate membrane trafficking in cells. Consistent with these findings, C9ORF72 localizes to endosomes, autophagosomes, and lysosomes, where it colocalizes with several Rab proteins [77, 78]. Furthermore, siRNA against C9ORF72 disrupts endocytosis and autophagy, leading to subcellular aggregation of p62 and/or TDP-43 in cultured neurons [77]. In agreement with these data, overexpressing C9ORF72 activates autophagy, as indicated by upregulated autophagosome formation. Interestingly, it is suggested that C9ORF72 activates autophagy via ULK1, a target protein of TBK1 [79, 80]. Since loss of TBK1 can also cause ALS and FTD [13], these data suggest that impaired autophagy may be a common pathogenic pathway in both C9ORF72- and TBK1-mediated ALS/FTD.
In agreement with the role of C9ORF72 in cellular vesicle trafficking, C9ORF72 KO mice exhibit endolysosomal and/or autophagic defects, but these defects vary across different tissues. In macrophages, C9ORF72 KO impairs autophagy and endolysosomal trafficking [45], whereas, in brain cells or fibroblasts, it increases autophagic flux [73]. In addition, C9ORF72 KO also suppresses mTOR signaling and increases the nuclear level of TFEB, a master regulator of lysosomal biogenesis, in fibroblasts [73], suggesting that C9ORF72 regulates autophagy at multiple steps in different tissues.
In summary, C9ORF72 functions as a RabGEF when complexing with other proteins, which regulates vesicle trafficking in cells. Its loss causes endolysosomal and autophagic defects both in vitro and in vivo. While insufficient to initiate neurodegeneration, C9ORF72 loss impairs autophagy, which may contribute to neurodegeneration when combined with the gain-of-toxicity mechanism.
Gain of toxicity
While the G4C2 HRE leads to loss of C9ORF72, most evidence suggests that it causes diseases via a gain-of-toxicity mechanism. Indeed, the HRE produces G4C2 and C4G2 repeat mRNAs, which are believed to be responsible for the toxicity. However, how repeat mRNAs cause toxicity is unclear. So far, three models have been proposed: 1) The repeat mRNAs can form guanine-quadruplex (G-quartet) secondary structures, which bind to RNA-binding proteins (RBPs) and lead to their loss of function [16, 17, 37]; 2) The repeat mRNAs can form hybrids called “R-loops” with DNA double strands, which cause DNA damage [81]; 3) The repeat-containing unspliced mRNAs [82, 83] or spliced intronic RNA [84] can undergo RAN translation to generate DPRs, which are toxic [22, 29, 34, 85, 86].
These three models are nonexclusive, but the first two argue that the repeat mRNAs cause cytotoxicity by their secondary structure, whereas the third one argues that they cause cytotoxicity through DPRs. Consistent with these models, both sense and antisense repeat RNA foci, R-loops, and aggregates of all five DPR species have been observed in C9ALS/FTD patient tissues and model systems [12, 18, 20, 21, 48, 81, 87, 88]. In addition, antisense oligonucleotides (ASOs) or knockdown of transcription elongation factors reduces the levels of both repeat RNAs and DPRs and suppresses HRE-mediated toxicity in multiple C9ALS/FTD model systems [16, 31, 38, 40, 71]. However, whether the repeat RNAs cause cytotoxicity via their secondary structures or DPRs is under debate.
Small molecules targeting the RNA secondary structures have been shown to suppress cytotoxicity or neurodegeneration in C9ALS/FTD models, but these molecules also reduce DPR levels [37, 89, 90]. To distinguish the effects of the RNA secondary structures from DPRs, Mizielinska et al. [33] have generated Drosophila models expressing G4C2 repeats interrupted by stop codons in all frames (the “RNA-only,” or “RO,” flies) [33]. These flies express mRNAs with G-quartet secondary structures, but no DPR, and do not undergo neurodegeneration, whereas flies expressing regular G4C2 repeats exhibit severe neurodegeneration, suggesting that the G-quartet is non-toxic. This idea is supported by studies in another C9ALS/FTD Drosophila model, which exhibits RNA foci, but no detectable DPR or overt neurodegeneration [34]. However, it is unclear whether the G-quartets from the “RO” RNA bind to the same RBPs as the regular repeat RNA or whether the “RO” RNA forms R-loops. Importantly, the evidence against secondary-structure-mediated toxicity has so far been limited to studies in Drosophila, whereas in Zebrafish, both sense and antisense “RO” RNAs have been shown to exert neuronal toxicity [64]. To better understand the role of the secondary structures of the repeat RNAs in C9ALS/FTD pathogenesis, future studies can test the “RO” constructs in mammalian models.
Despite the controversy over whether the RNA secondary structures play a role in pathogenesis, it is generally agreed that DPRs are cytotoxic. Many studies have shown that DPRs encoded by alternative codons(i.e. non-HRE) cause toxicity in multiple model systems (reviewed in [24]). In addition, genetically or pharmacologically inhibiting DPR translation suppresses neuronal defects in C9ALS/FTD models, without affecting RNA foci [31, 33, 39, 91]. While the overall DPR pathology does not correlate with the affected brain regions in patients [88, 92,93,94], poly(GR) pathology does [95, 96]. Together, these studies suggest a critical role of DPRs in pathogenesis.
Among all five DPR species, the arginine-containing DPRs, i.e. poly(GR) and poly(PR), are the most toxic. In fly models, poly(GR) and poly(PR), but not other DPR species, cause neurodegeneration [33]. Consistent with these findings, poly(GR) and poly(PR) exhibit cytotoxicity in other model systems, including yeast, worm, mouse, and cultured cells, whereas neither poly(GP) nor poly(PA) is toxic in cultured cells [32, 49, 51, 62, 85, 97, 98]. For poly(GA), some studies reported its toxicity in mice and cultured neurons [50, 86, 99, 100]. However, others reported little or no poly(GA) toxicity in flies or cultured neurons, whereas poly(GR) and/or poly(PR) expressed in the same systems exhibit strong toxicity [29, 33, 98]. Thus, poly(GA) can be toxic under certain conditions but is much less toxic compared to poly(GR) and poly(PR), when these DPRs are expressed at similar levels. In agreement with these findings, mass spectrometry analyses have identified more than 200 endogenous proteins to specifically interact with poly(GR) and poly(PR) when these DPRs are overexpressed in cultured cells, whereas only six proteins specifically interact with poly(GA), and none specifically interact with poly(GP) and poly(PA) [36].
Although the repeat RNAs and some DPR species cause little or no toxicity when expressed alone, it is possible that they can enhance poly(GR)- or poly(PR)-mediated cytotoxicity. For example, some RNA has been shown to promote liquid-liquid phase separation (LLPS) of poly(GR) and poly(PR), a process associated with poly(GR)- and poly(PR)-mediated cytotoxicity [101]. Furthermore, poly(GA) and poly(GP) sometimes co-aggregate with poly(GR) and/or poly(PR), suggesting that they may enhance the aggregation propensity of poly(GR) and poly(PR) [86].
Despite the debate over the RNA-secondary-structure- versus DPR-mediated toxicities, recent studies have suggested that they converge on the same downstream, functional defects in cells, including nucleocytoplasmic transport disruption, membrane-less organelle defects, and DNA damage (Figure 2). Indeed, emerging evidence has suggested the importance of these defects in pathogenesis, which will be the focus of this review. In addition, we will also discuss other functional defects implicated in C9ALS/FTD.

Connections among functional defects downstream of repeat RNAs and DPRs. a G-quartets formed by repeat RNAs disrupt nucleocytoplasmic transport by sequestering RanGAP. It also promotes stress granule assembly. b DPRs induce stress granule assembly, which disrupts nucleocytoplasmic transport, inhibits global translation, and enhances RAN translation. Enhanced RAN translation produces more DPRs, forming a feedforward loop. c G-quartets formed by repeat RNAs disrupt nucleoli by sequestering NCL, whereas DPRs disrupt nucleoli by co-phase separating with NPM1 and NCL. In addition, DPRs cause DNA damage via NPM1 and nucleolar defects. d Repeat RNAs form R-loops, which causes DNA damage. e Cytoplasmic DPRs disrupt mitochondrial function, producing ROS that causes DNA damage. These discoveries not only provide us with an integrated view of C9ALS/FTD pathogenic mechanism but also help us better understand the fundamental biology underlying neurodegeneration.
Nucleocytoplasmic Transport Disruption
Several studies have independently identified a critical role of nucleocytoplasmic transport disruption in C9ALS/FTD, which can stem from either RNA secondary structure or DPRs [28, 29, 32, 37, 102]. One study from us has shown that the G-quartets formed by the sense repeat RNA bind to and sequester RanGAP1, a key regulator of nucleocytoplasmic transport, which impairs the nuclear import of proteins in C9ALF/FTD fly and iPSN models [37]. Importantly, a nuclear export inhibitor, KPT-276, restores the balance between import and export and suppresses neurodegeneration in the fly model, suggesting nucleocytoplasmic transport defects as a potential therapeutic target. In parallel to this study, another study has shown that the sense repeat RNA and/or arginine-containing DPRs disrupt the nuclear export of mRNAs in another C9ALS/FTD fly model [29]. Consistent with these studies, genetic screens have implicated nucleocytoplasmic transport disruption in yeast or flies expressing poly(PR) or C9ALS/FTD patient iPSNs [28, 32]. The importance of nucleocytoplasmic transport disruption is further supported by immunohistochemistry staining in C9ALS/FTD patient or mouse tissues, which exhibit aggregation of nucleocytoplasmic transport factors [37, 47, 50, 51].
The idea that the G-quartets disrupt nucleocytoplasmic transport by binding to RanGAP1 [37] is further supported by a large-scale interactome study identifying RanGAP1 as an interactor of GC-enriched RNAs [103]. However, how DPRs disrupt nucleocytoplasmic transport is under debate. Shi et al. [102] have shown that a chemically synthesized poly(PR) peptide blocks nuclear pore by binding to nucleoporins, leading to nucleocytoplasmic transport defects [102]. In contrast, a recent study has failed to detect similar defects induced by the same peptide [104], possibly due to different experimental settings in these two studies. The second study challenges the idea that DPRs directly block nuclear pores, raising the possibility that DPRs disrupt nucleocytoplasmic transport through an indirect mechanism.
The discovery of DPRs disrupting nucleocytoplasmic transport has triggered a surge of further studies, which have identified nucleocytoplasmic transport disruption as a common cellular defect in protein-misfolding diseases [105,106,107,108]. Upon protein misfolding stress, cells halt their translation by embedding their mRNAs in large RNA/protein condensates called stress granules, in which many nucleocytoplasmic transport factors are sequestered. As DPRs have been shown to induce stress granule assembly in some cells, it is likely that DPRs disrupt nucleocytoplasmic transport by sequestering transport factors in stress granules. In agreement with this hypothesis, we have shown that many transport factors localize to DPR-induced stress granules and stress granule inhibitors partially suppress nucleocytoplasmic transport defects in cells overexpressing DPRs [109]. Furthermore, Vanestte et al. (2019) have observed a correlation between the presence of stress granules and nucleocytoplasmic transport defects in cells expressing DPRs at low levels [104]. Together, these findings suggest that DPRs can disrupt nucleocytoplasmic transport through stress granule assembly.
Membrane-less Organelle Defects
Stress granules are a group of membrane-less organelles, subcellular compartments that lack surrounding lipid membranes and perform certain biochemical functions. These organelles are protein/RNA condensates enriched in proteins with low-complexity domains (LCDs). Many LCD-containing proteins are aggregate-prone and can undergo LLPS, a process in which molecules demix in an aqueous environment. It has been shown that proper LLPS plays a key role in the assembly, dynamics, and functions of membrane-less organelles, whereas aberrant LLPS can cause aggregation of LCD-containing proteins, as well as defects in these organelles (reviewed in [110]).
In C9ALS/FTD, both the sense repeat RNA and DPRs have been shown to disrupt membrane-less organelles, including stress granules and nucleoli. Indeed, poly(GR) and poly(PR) interactomes are enriched in protein components of stress granules, nucleoli, and other membrane-less organelles, and many of these proteins also genetically interact with poly(GR) or poly(PR) [36, 111]. Furthermore, in vivo studies have suggested an adverse effect of poly(PR) on heterochromatin protein complexes [51], another form of membrane-less organelles. We will discuss these studies in this section.
-
a)
Stress Granule
Stress granules are cytoplasmic RNA/RBP condensates, but certain stimuli can convert them into protein aggregates. Interestingly, several ALS/FTD proteins, including TDP-43, FUS, Ataxin-2, TIA1, and hnRNPs, are stress granule proteins. Since the cytoplasmic aggregation of TDP-43, FUS, and hnRNPs is a pathological hallmark of ALS/FTD, including C9ALS/FTD, stress granules are believed the key to ALS/FTD pathogenesis. So far, this idea has been supported by genetic, cell biology, and pathological evidence in many model systems or patients (reviewed in [7]). However, other studies have shown that TDP-43 can aggregate in cultured cells via stress-granule-independent routes [112, 113]. Furthermore, some TDP-43 aggregates in ALS/FTD model systems or patients do not colocalize with stress granule markers [114, 115], suggesting that stress granules are insufficient to cause all TDP-43 pathology in ALS/FTD. Besides its role in triggering protein aggregation, stress granule assembly inhibits global translation and disrupts nucleocytoplasmic transport [82, 84, 109, 116], which can contribute to neurodegeneration. Thus, stress granules contribute to ALS/FTD pathogenesis through multiple mechanisms.
In C9ALS/FTD, both the G4C2 repeat RNA and DPRs promote stress granule assembly. The G-quartets formed by the G4C2 repeat RNA bind to and condense RBPs that are stress granule components, promoting RBP LLPS. Consistent with these findings, cells transfected with synthetic G4C2 repeat RNA exhibit stress granules, which occasionally colocalize with the repeat RNAs [117]. However, it is unclear whether these stress granules are caused by G-quartets or DPRs. Indeed, all five DPRs have been shown to induce stress granule assembly without additional stress [36, 98, 101, 109, 118]. Importantly, poly(GR) and poly(PR) localize to stress granules that they induce, bind to, alter the LLPS propensity, and impair the dynamics of stress granule proteins [36, 101]. As the dynamics of proteins negatively correlates with their propensity to aggregate, these studies suggest that poly(GR) and poly(PR) promote the aggregation of stress granule proteins. Indeed, poly(GR) co-aggregates with several stress granule proteins in the brain of C9ALS/FTD patients and mouse models [47, 49], suggesting that poly(GR) causes aggregation of stress granule proteins in vivo. Consistent with the role of stress granules in translation repression, poly(GR) and poly(PR) have been shown to suppress global translation in multiple model systems [36, 49, 104, 119, 120].
In accord with these mechanistic studies, C9ALS/FTD iPSNs exhibit an increased propensity to form, and a decreased rate to clear, stress granules [121]. Importantly, inhibiting stress granule assembly by either knockdown of genes or chemical inhibitors suppresses neurodegeneration in C9ALS/FTD iPSN and fly models [109], suggesting the potential of stress granules as a therapeutic target.
-
b)
Nucleolus
Nucleoli are nuclear RNA/RBP condensates where ribosomal biogenesis occurs. They contain many proteins that undergo LLPS, such as NCL and NPM1. Both G4C2 repeat RNA and arginine-containing DPRs have been shown to disrupt nucleolar structure and function. Haeusler et al. [17] have shown that the G-quartets formed by G4C2 repeat RNA bind to NCL, causing NCL mislocalization and nucleolar stress [17]. Consistent with these data, nucleoli are expanded in cultured lymphocytes from C9ALS/FTD patients, and the nucleolar function is impaired in patient motor cortices. Together, these findings suggest that repeat RNAs disrupt nucleolar structure and function. While Haeusler et al. [17] reported expanded nucleoli in C9ALS/FTD lymphocytes, another study has observed no nucleolar size change in postmortem brains [88]. However, the results from the latter study could be due to pathological heterogeneity in brain cells. Indeed, a third study has shown that the nucleolar size is increased in C9ALS/FTD postmortem brains if only cells with repeat RNA foci are quantified [122], suggesting that the repeat RNAs indeed increase the nucleolar size. Interestingly, this study has also shown that brain cells without poly(GR) aggregation exhibit reduced nucleolar size, whereas cells containing poly(GR) aggregation exhibit enlarged nucleoli [122], suggesting a role of poly(GR) in nucleolar stress.
Several studies have shown that either overexpressed or chemically synthesized poly(GR) and poly(PR) localize to nucleoli and disrupt nucleolar functions [36, 85, 98, 101, 118, 123]. Furthermore, poly(GR) and poly(PR) undergo LLPS and bind to many nucleolar proteins, including NCL and NPM1, which disturbs LLPS and impair the dynamics and function of NCL and NPM1 [36, 101, 111, 123]. Together, these data suggest that poly(GR) and poly(PR) disrupt nucleolar function in cultured cells by impairing LLPS of key nucleolar proteins.
Despite their nucleolar toxicity in cultured cells, poly(GR) and poly(PR) rarely localize to nucleoli in C9ALS/FTD patient and mouse models and fail to induce nucleolar stress when overexpressed in mouse brains [49, 51, 88]. To define the precise role of poly(GR) and poly(PR) in nucleolar function in vivo, future studies may focus on whether they disrupt nucleoli when combined with the repeat RNAs and/or other DPRs in mouse models.
-
c)
Heterochromatin Protein Complex
In a C9ALS/FTD mouse model overexpressing poly(PR), Zhang et al. [51] have unexpectedly discovered that poly(PR) rarely localizes to nucleoli [51], despite previous findings in cultured cells. Interestingly, poly(PR) mostly localizes to heterochromatin, DNA/protein condensates containing transcriptionally silent chromatin, in these mice. The authors have also discovered that poly(PR) localizes to heterochromatin in some C9ALS/FTD patient neurons. Consistent with these findings, transcripts from heterochromatin accumulate in both these mice and patients, suggesting heterochromatin defects.
Heterochromatin formation is mediated by heterochromatin protein 1 (HP1) proteins, which bind to chromatin and recruit other chromatin-interacting proteins. Recent studies have suggested that the heterochromatin compartment is a membrane-less organelle formed by LLPS of HP1α, an HP1 protein that contains LCDs [124, 125]. Interestingly, poly(PR) disrupts HP1α LLPS and reduces HP1α levels in mice overexpressing poly(PR) [51], suggesting a mechanism by which poly(PR) disrupts heterochromatin. These findings and previous findings in cultured cells [36, 101, 111] have converged on the adverse effects of poly(PR) on LLPS of LCD-containing proteins.
-
d)
Other Membrane-less Organelles
In addition to stress granule and nucleolar proteins, components of other membrane-less organelles, including nuclear speckles and Cajal bodies, have been identified as physical and/or genetic interactors of poly(GR) and/or poly(PR) [36, 111]. Furthermore, these DPRs also alter nuclear speckle dynamics and Cajal body assembly in cultured cells, possibly through similar mechanisms as they disrupt stress granules and nucleoli.
In summary, both the G4C2 repeat RNA and DPRs, especially poly(GR) and poly(PR), disrupt multiple types of membrane-less organelles both in vivo and in vitro by binding to and impairing LLPS of proteins in these organelles. Notably, both the G4C2 repeat RNA and arginine-containing DPRs have been shown to undergo LLPS, but whether their LLPS propensity correlates with their toxicity on membrane-less organelles is unclear. Nevertheless, it has been shown that poly(GR) co-aggregates with stress granule proteins in both C9ALS/FTD mouse and patient neurons [51, 109]. Thus, it is intriguing to hypothesize that LLPS triggers the aggregation of repeat RNAs, arginine-containing DPRs, and their interactors in membrane-less organelles.
DNA Damage
Besides impaired nucleocytoplasmic transport and membrane-less organelles, DNA damage can also be caused by either repeat RNAs or DPRs and has been observed in multiple model systems of C9ALS/FTD, as well as in patients [81, 97, 126, 127]. So far, studies have identified several routes by which the repeat RNAs or DPRs can cause DNA damage. Firstly, the repeat RNAs cause DNA double-strand breaks via R-loops. Secondly, the repeat RNAs and poly(GA) cause DNA damage by disrupting the ataxia-telangiectasia-mutated-signaling pathway, a critical component in DNA damage repair, and/or reducing the level of hnRNP A3 [81, 127]. Thirdly, poly(GR) interacts with mitochondrial proteins and disrupts mitochondrial function, leading to increased oxidative stress and DNA damage in iPSNs [126]. As DNA damage in these iPSNs can be partially suppressed by antioxidants, it is suggested that poly(GR) causes DNA damage partially through oxidative stress [126, 128]. Fourthly, poly(GR) and poly(PR) may cause DNA damage by disrupting the function of NPM1, a nucleolar protein that functions in DNA damage repair [97]. These mechanisms are non-exclusive and likely co-exist in patients. Notably, several proteins involved in DNA damage repair, e.g. NPM1, TDP-43, and FUS, undergo LLPS and are known to interact with poly(GR) and poly(PR). Thus, future studies may test the possibility that poly(GR) and poly(PR) damage DNA by disrupting LLPS of these proteins.
Although DNA damage is generally believed to cause neurodegeneration, direct evidence is lacking to demonstrate that it contributes to C9ALS/FTD pathogenesis. Resolving R-loops by Senataxin or suppressing oxidative stress by SOD1 and/or catalase ameliorates repeat-RNA- and/or DPR-mediated toxicity [81, 126]. However, these approaches target factors upstream of DNA damage. Given the technical challenges to modulate DNA damage directly, future studies can focus on up- and downstream factors of DNA damage.
Other Functional Defects
In addition to nucleocytoplasmic transport disruption, membrane-less organelle defects, and DNA damage, many other functional defects have been implicated in C9ALS/FTD. For example, poly(GR) has been shown to disrupt mitochondrial function by sequestering key mitochondrial proteins in iPSN and/or mouse models [126, 128]. Consistent with these findings, mitochondrial morphology is disrupted in C9ALS/FTD fibroblasts [129]. Poly(GR) and poly(PR) have also been shown to disrupt axonal transport of mitochondria and vesicles [130]. Furthermore, poly(GA) has been shown to impair the proteasome system [50, 131]. In addition, dysregulation of RNA editing has been implicated in C9ALS/FTD [132]. For a list of functional defects implicated in C9ALS/FTD, please see Table 1. These findings suggest the complex pathophysiology of C9ALS/FTD, with a remaining question as to how these defects connect to each other.
Connections among Functional Defects
Eukaryotic cells, including neurons, are highly ordered, dynamic entities with intricate organization. Defects in one cellular organelle or process usually disrupt others. Indeed, many cellular functional defects in C9ALS/FTD are connected. Stress granule assembly has been shown to disrupt nucleocytoplasmic transport in C9ALS/FTD by sequestering transport factors in stress granules [109], whereas disrupted nucleocytoplasmic transport may cause RNA editing enzyme ADAR2 to mislocalize to the cytoplasm, which causes RNA editing defects in C9ALS/FTD [132]. In addition, mitochondrial defects have been shown to cause DNA damage in C9ALS/FTD via reactive oxygen species [126], whereas nucleolar defects may also contribute to DNA damage in C9ALS/FTD via disrupted NPM1 function [97]. Furthermore, DPR-induced stress granule assembly inhibits global translation but selectively enhances RAN translation, thereby promoting DPR production [82, 84, 116]. These findings have not only revealed molecular cascades and feedforward mechanisms underlying the pathogenesis but also help us better understand the intricate organization of eukaryotic cells—a fundamental issue in cell biology.
Therapeutic Development
Since the major cause of C9ALS/FTD appears to be the gain of toxicity derived from the repeat RNAs and their translational products, DPRs, methods to eliminate these toxic species have been actively explored. Particularly, an ASO therapy is more advanced than other therapies in clinical development. Notably, the repeat RNAs and DPRs are toxic because they disrupt certain function(s) of cells. Thus, targeting functional defects downstream of the repeat RNAs and/or DPRs holds great therapeutic potential. Indeed, a chemical compound targeting nucleocytoplasmic transport, which stems from our discoveries [37], is currently in an ALS clinical trial. In this section, we will review the current progress on C9ALS/FTD therapeutic development and suggest potential ideas for future studies.
Targeting repeat RNAs and/or DPRs
C9ORF72 ASO
ASOs are synthetic single-stranded oligonucleotides that can bind to their target RNAs with very high specificity. This binding can restore, modify, inactivate, or promote the degradation of the RNAs. ASOs are stable and effective in the central nervous system, and an ASO modifying gene expression has been used to successfully treat spinal muscular atrophy, another motor neuron degenerative disease [133,134,135,136]. In addition, an ASO that activates RNase-H-mediated mRNA degradation has exhibited promising effects on patients with SOD1-related ALS (ALS1) and is now in a phase III study (https://clinicaltrials.gov/ct2/show/NCT02623699?term=Tofersen&draw=2&rank=1), supporting the efficacy of ASOs in treating ALS caused by a gain-of-toxicity mutation.
For C9ALS/FTD, current ASO therapies focus on degrading the sense transcripts. The ASOs against C9ORF72 have been shown to suppress pathological and pathophysiological defects, as well as neurodegeneration, in multiple model systems [16, 37, 71, 137,138,139]. Phase I clinical studies with an ASO that activate RNase-H-mediated degradation of the sense transcripts are underway through Ionis and Biogen (https://clinicaltrials.gov/ct2/show/NCT03626012).
Despite its promise, there are concerns related to ASO therapy. First, the current ASO used in clinical studies does not directly impact the antisense transcript, which may leave part of the pathology unaffected. Furthermore, SMA and ALS1 mainly affect lower motor neurons, whereas C9ALS/FTD is often related to strong upper-motor-neuron affliction. To treat C9ALS/FTD, the pharmacokinetics and pharmacodynamics of ASOs in upper motor neurons must be determined. It is also worth noting that ASOs treating SMA and ALS have different effects on mRNAs—the ASO for SMA blocks an internal splicing site whereas the ASOs for ALS1 and C9ALS/FTD degrade mRNAs. Thus, the clinical effects of these ASOs may be different.
Other Molecules
Small molecules modulating G4C2 RNA G-quartets or antibodies against poly(GA) have been shown to suppress HRE-mediated toxicity in cell and/or Drosophila models of C9ALS/FTD [37, 90, 140]. Future studies may focus on their effects in mammals. Furthermore, Metformin, an FDA-approved drug to treat diabetes, has been shown to reduce RAN translation. An ongoing clinical study is assessing its safety and tolerability in C9ALS/FTD patients (https://clinicaltrials.gov/ct2/show/NCT04220021).
In addition to the ASOs and molecules discussed above, proteins implicated in the production of repeat RNAs or DPRs [30, 38, 91] may also be therapeutic targets. Furthermore, AAV-mediated delivery of gene-silencing tools, such as CRISPR/Cas9, is in its infancy. Future research can test these approaches.
Targeting Downstream Defects
Nuclear Export Inhibitor KPT-350 (BIIB100)
A pathological hallmark of ALS/FTD, including C9ALS/FTD, is cytoplasmic mislocalization of TDP-43 [8, 9], potentially due to impaired nuclear import. Thus, inhibiting nuclear export may restore the nuclear import/export balance and suppress neurodegeneration. Indeed, we have shown that an Exportin-1 inhibitor, KPT-276, suppresses nucleocytoplasmic transport defects, as well as neurodegeneration, in a C9ALS/FTD fly model [37]. In addition, another Exportin-1 inhibitor, KPT-350, a.k.a. BIIB100, suppresses neurodegeneration in a rat model of TDP-43-mediated ALS/FTD [141], suggesting its therapeutic potential. Currently, a phase I clinical study on KPT-350 is underway (https://clinicaltrials.gov/ct2/show/NCT03945279). However, recent studies have suggested that the nuclear export of TDP-43 is not mediated by Exportin-1 [142, 143] and KPT-350 does not suppress TDP-43 mislocalization in the ALS/FTD rat model [141], raising the question as to how KPT-350 suppresses neurodegeneration. Interestingly, KPT-350 has been shown to suppress inflammatory responses in the central nervous system [144], possibly because Exportin-1 exports transcription factors implicated in anti-inflammatory responses from the nucleus. Hence, it is possible that inflammatory responses contribute to C9ALS/FTD pathogenesis.
Stress Granule Inhibitors and Ataxin-2 ASO
Stress granule assembly is believed to contribute to ALS/FTD pathogenesis by triggering the aggregation of TDP-43, FUS, and other hnRNPs. Consistent with this hypothesis, inhibiting stress granule assembly using chemical inhibitors or ASOs against Ataxin-2, an essential stress granule component, has been shown to suppress defects in iPSN and/or Drosophila models of C9ALS/FTD [109]. Stress granule inhibitor 2BAct has also been shown neuroprotective in a mouse model of Vanishing White Matter, another neurodegenerative disease [145]. In addition, Ataxin-2 ASOs have been shown to suppress neurodegeneration in mouse models of TDP-43-related ALS or spinocerebellar ataxia 2 [146, 147]. Together, these findings have suggested the therapeutic potential of these approaches. Recently, a high-content drug screen has identified ~100 small molecules that modulate stress granules in cultured cells [148], providing a large pool of candidate drugs for future in vivo studies.
In addition to nucleocytoplasmic transport and stress granules, other downstream, functional defects may also be therapeutic targets. Importantly, given that some of these defects also present in other types of ALS or FTD, strategies targeting these defects are likely able to be translated from C9ALS/FTD to other ALS/FTD cases. For example, the clinical trial on KPT-350 also includes sporadic ALS patients. However, before targeting a downstream defect, one must carefully evaluate whether this defect is indeed the cause, rather than simply a consequence or byproduct, of neurodegeneration. Thus, rescuing with genetic and pharmacological manipulations in model systems is highly recommended before further analyses.
Conclusions
Like many other neurodegenerative diseases, C9ALS/FTD has a complex pathophysiology. Although it is widely accepted that the G4C2 HRE causes neurodegeneration via a gain of toxicity, emerging evidence suggests that loss of C9ORF72 also plays an auxiliary role. Furthermore, both the loss and gain of function cause defects in many subcellular organelles/processes that contribute to C9ALS/FTD neurodegeneration. Importantly, some of these defects also occur in other forms of ALS/FTD and other neurodegenerative diseases (e.g. Alzheimer’s disease), as well as during cellular responses to a variety of stress. Thus, these studies also help us better understand not only the mechanisms of other diseases but also the principles of fundamental cell biology.
Despite recent progress, several questions remain unanswered. Firstly, some studies suggest the involvement of Golgi and the extracellular matrix in pathogenesis with the underlying mechanism unclear. Secondly, how glia contributes to the diseases is ill-defined. Importantly, it remains unclear whether the downstream defects caused by G4C2 HRE occur in parallel or in series. Identifying defects that occur early in pathogenesis is crucial, as targeting these events will likely yield clinical effects.
If the ASO approach will treat C9ALS/FTD in the future, as it does for SMA, one may question the value of studying downstream cellular defects. However, many of these defects also occur in sporadic ALS or FTD cases, which lack an obvious genetic cause. Thus, even from a clinical perspective, understanding the cellular and molecular basis of these diseases is still vital.
Availability of data and materials
Not applicable.
Change history
01 July 2020
An amendment to this paper has been published and can be accessed via the original article.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- FTD:
-
Frontotemporal dementia
- TDP-43:
-
TAR-DNA binding protein 43
- FUS:
-
Fused in sarcoma
- HnRNP:
-
Heterogeneous nuclear ribonucleoproteins
- C9ORF72:
-
Chromosome 9 open reading frame 72
- G4C2 :
-
GGGGCC
- HRE:
-
Hexanucleotide repeat expansion
- RAN:
-
Repeat-associated, non-ATG
- DPR:
-
Dipeptide repeat protein
- C9ALS/FTD:
-
C9ORF72-mediated ALS and FTD
- iPSN:
-
Neurons derived from induced pluripotent stem cells
- AAV:
-
Adeno-associated-virus
- LLPS:
-
Liquid-liquid phase separation
- LCD:
-
Low-complexity domain
- RBP:
-
RNA-binding protein
- ASO:
-
Antisense oligonucleotide
References
Ryan M, Heverin M, McLaughlin RL, Hardiman O. Lifetime Risk and Heritability of Amyotrophic Lateral Sclerosis. JAMA Neurol. 2019;76(11):1367-74.
Young JJ, Lavakumar M, Tampi D, Balachandran S, Tampi RR. Frontotemporal dementia: latest evidence and clinical implications. Ther Adv Psychopharmacol. 2018;8:33–48.
Ferrari R, Kapogiannis D, Huey ED, Momeni P. FTD and ALS: a tale of two diseases. Curr Alzheimer Res. 2011;8:273–94.
Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–54.
Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–73.
Kwiatkowski TJ Jr, Bosco DA, LeClerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–8.
Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol. 2013;201:361–72.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3.
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–11.
Bahia VS, Takada LT, Deramecourt V. Neuropathology of frontotemporal lobar degeneration: a review. Dement Neuropsychol. 2013;7:19–26.
Bannwarth S, Ait-El-Mkadem S, Chaussenot A, Genin EC, Lacas-Gervais S, Fragaki K, Berg-Alonso L, Kageyama Y, Serre V, Moore DG, et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain. 2014;137:2329–45.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–56.
Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K, Marroquin N, Nordin F, Hubers A, Weydt P, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18:631–6.
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–68.
Rubino E, Rainero I, Chio A, Rogaeva E, Galimberti D, Fenoglio P, Grinberg Y, Isaia G, Calvo A, Gentile S, et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012;79:1556–62.
Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–28.
Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200.
Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, DeJesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW III, Rademakers R, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–46.
Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PE, Caulfield T, Daughrity L, Dunmore JH, Castanedes-Casey M, Chew J, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013;126(6):829-44.
Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K, Weng SM, Schludi MH, van der Zee J, Cruts M, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013a;126:881–93.
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van BC, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013b;339:1335–8.
Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A. 2013;110:E4968–77.
Balendra R, Isaacs AM. C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol. 2018;14:544–58.
Freibaum BD, Taylor JP. The Role of Dipeptide Repeats in C9ORF72-Related ALS-FTD. Front Mol Neurosci. 2017;10:35.
Gitler AD, Tsuiji H. There has been an awakening: Emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res. 2016;1647:19–29.
Haeusler AR, Donnelly CJ, Rothstein JD. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci. 2016;17:383–95.
Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–38.
Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovicic A, De BG, Scheveneels W, Steyaert J, Cuijt I, et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep. 2016;6:20877.
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525:129–33.
Goodman LD, Prudencio M, Kramer NJ, Martinez-Ramirez LF, Srinivasan AR, Lan M, Parisi MJ, Zhu Y, Chew J, Cook CN, et al. Toxic expanded GGGGCC repeat transcription is mediated by the PAF1 complex in C9orf72-associated FTD. Nat Neurosci. 2019a;22:863–74.
Goodman LD, Prudencio M, Srinivasan AR, Rifai OM, Lee VM, Petrucelli L, Bonini NM. eIF4B and eIF4H mediate GR production from expanded G4C2 in a Drosophila model for C9orf72-associated ALS. Acta Neuropathol Commun. 2019b;7:62.
Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW III, Sun S, Herdy JR, Bieri G, et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 2015;18:1226–9.
Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IO, Pietrzyk J, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345:1192–4.
Tran H, Almeida S, Moore J, Gendron TF, Chalasani U, Lu Y, Du X, Nickerson JA, Petrucelli L, Weng Z, et al. Differential Toxicity of Nuclear RNA Foci versus Dipeptide Repeat Proteins in a Drosophila Model of C9ORF72 FTD/ALS. Neuron. 2015;87:1207–14.
Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS, et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A. 2013;110:7778–83.
Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, Messing J, Molliex A, et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell. 2016;167:774–88.
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61.
Kramer NJ, Carlomagno Y, Zhang YJ, Almeida S, Cook CN, Gendron TF, Prudencio M, Van Blitterswijk M, Belzil V, Couthouis J, et al. Spt4 selectively regulates the expression of C9orf72 sense and antisense mutant transcripts. Science. 2016;353:708–12.
Yamada SB, Gendron TF, Niccoli T, Genuth NR, Grosely R, Shi Y, Glaria I, Kramer NJ, Nakayama L, Fang S, et al. RPS25 is required for efficient RAN translation of C9orf72 and other neurodegenerative disease-associated nucleotide repeats. Nat Neurosci. 2019;22:1383–8.
Yuva-Aydemir Y, Almeida S, Krishnan G, Gendron TF, Gao FB. Transcription elongation factor AFF2/FMR2 regulates expression of expanded GGGGCC repeat-containing C9ORF72 allele in ALS/FTD. Nat Commun. 2019;10:5466.
Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, Siao CJ, Brydges S, LaRosa E, Bai Y, et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016;6:23204.
Burberry A, Suzuki N, Wang JY, Moccia R, Mordes DA, Stewart MH, Suzuki-Uematsu S, Ghosh S, Singh A, Merkle FT, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med. 2016;8:347ra393.
Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, Yachnis AT, Ranum LP. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron. 2016;90:521–34.
O'Rourke JG, Bogdanik L, Muhammad AK, Gendron TF, Kim KJ, Austin A, Cady J, Liu EY, Zarrow J, Grant S, et al. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron. 2015;88:892–901.
O'Rourke JG, Bogdanik L, Yanez A, Lall D, Wolf AJ, Muhammad AK, Ho R, Carmona S, Vit JP, Zarrow J, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016;351:1324–9.
Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J, Weiss A, Wightman N, Salameh J, Kim J, et al. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron. 2015;88:902–9.
Chew J, Cook C, Gendron TF, Jansen-West K, Del Rosso G, Daughrity LM, Castanedes-Casey M, Kurti A, Stankowski JN, Disney MD, et al. Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol Neurodegener. 2019;14:9.
Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Lee CW, Jansen-West K, Kurti A, Murray ME, et al. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015;348:1151–4.
Zhang YJ, Gendron TF, Ebbert MTW, O'Raw AD, Yue M, Jansen-West K, Zhang X, Prudencio M, Chew J, Cook CN, et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat Med. 2018b;24:1136–42.
Zhang YJ, Gendron TF, Grima JC, Sasaguri H, Jansen-West K, Xu YF, Katzman RB, Gass J, Murray ME, Shinohara M, et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci. 2016;19:668–77.
Zhang YJ, Guo L, Gonzales PK, Gendron TF, Wu Y, Jansen-West K, O'Raw AD, Pickles SR, Prudencio M, Carlomagno Y, et al. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science. 2019;363(6428):eaav2606.
Cudkowicz ME, van den Berg LH, Shefner JM, Mitsumoto H, Mora JS, Ludolph A, Hardiman O, Bozik ME, Ingersoll EW, Archibald D, et al. Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): a randomised, double-blind, phase 3 trial. Lancet Neurol. 2013;12:1059–67.
de Carvalho M, Pinto S, Costa J, Evangelista T, Ohana B, Pinto A. A randomized, placebo-controlled trial of memantine for functional disability in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010;11:456–60.
Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C, Hilton JF, Spitalny GM, MacArthur RB, Mitsumoto H, et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. The Lancet Neurology. 2007;6:1045–53.
Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–21.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76.
Kramer NJ, Haney MS, Morgens DW, Jovicic A, Couthouis J, Li A, Ousey J, Ma R, Bieri G, Tsui CK, et al. CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat Genet. 2018;50:603–12.
Egawa N, Kitaoka S, Tsukita K, Naitoh M, Takahashi K, Yamamoto T, Adachi F, Kondo T, Okita K, Asaka I, et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci Transl Med. 2012;4:145ra104.
Tian R, Gachechiladze MA, Ludwig CH, Laurie MT, Hong JY, Nathaniel D, Prabhu AV, Fernandopulle MS, Patel R, Abshari M, et al. CRISPR Interference-Based Platform for Multimodal Genetic Screens in Human iPSC-Derived Neurons. Neuron. 2019;104:239–55 e212.
Ciura S, Lattante S, Le BI, Latouche M, Tostivint H, Brice A, Kabashi E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol. 2013;74:180–7.
Corrionero A, Horvitz HR. A C9orf72 ALS/FTD Ortholog Acts in Endolysosomal Degradation and Lysosomal Homeostasis. Curr Biol. 2018;28:1522–35 e1525.
Rudich P, Snoznik C, Watkins SC, Monaghan J, Pandey UB, Lamitina ST. Nuclear localized C9orf72-associated arginine-containing dipeptides exhibit age-dependent toxicity in C. elegans. Hum Mol Genet. 2017;26:4916–28.
Swaminathan A, Bouffard M, Liao M, Ryan S, Callister JB, Pickering-Brown SM, Armstrong GAB, Drapeau P. Expression of C9orf72-related dipeptides impairs motor function in a vertebrate model. Hum Mol Genet. 2018;27:1754–62.
Swinnen B, Bento-Abreu A, Gendron TF, Boeynaems S, Bogaert E, Nuyts R, Timmers M, Scheveneels W, Hersmus N, Wang J, et al. A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol. 2018;135:427–43.
Therrien M, Rouleau GA, Dion PA, Parker JA. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS One. 2013;8:e83450.
Belzil VV, Bauer PO, Prudencio M, Gendron TF, Stetler CT, Yan IK, Pregent L, Daughrity L, Baker MC, Rademakers R, et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013;126:895–905.
Fratta P, Poulter M, Lashley T, Rohrer JD, Polke JM, Beck J, Ryan N, Hensman D, Mizielinska S, Waite AJ, et al. Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol. 2013;126:401–9.
Gijselinck I, Van LT, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van CC, Pereson S, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11:54–65.
van Blitterswijk M, Gendron TF, Baker MC, DeJesus-Hernandez M, Finch NA, Brown PH, Daughrity LM, Murray ME, Heckman MG, Jiang J, et al. Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol. 2015;130:863–76.
Waite AJ, Baumer D, East S, Neal J, Morris HR, Ansorge O, Blake DJ. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging. 2014;35:1779.
Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, Stauffer JE, Jafar-Nejad P, Drenner K, Schulte D, et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron. 2016;90:535–50.
Koppers, M., Blokhuis, A.M., Westeneng, H.J., Terpstra, M.L., Zundel, C.A., Vieira de, S.R., Schellevis, R.D., Waite, A.J., Blake, D.J., Veldink, J.H., et al. (2015). C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol 78, 426-438.
Ugolino J, Ji YJ, Conchina K, Chu J, Nirujogi RS, Pandey A, Brady NR, Hamacher-Brady A, Wang J. Loss of C9orf72 Enhances Autophagic Activity via Deregulated mTOR and TFEB Signaling. PLoS Genet. 2016;12:e1006443.
Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, Hendricks E, Linares GR, Wang Y, Son EY, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 2018;24:313–25.
Zhu Q, Jiang J, Gendron TF, McAlonis-Downes M, Jiang L, Taylor A, Diaz Garcia S, Ghosh Dastidar S, Rodriguez MJ, King P, et al. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat Neurosci. 2020;23:615–24.
Sellier C, Campanari ML, Julie Corbier C, Gaucherot A, Kolb-Cheynel I, Oulad-Abdelghani M, Ruffenach F, Page A, Ciura S, Kabashi E, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016;35:1276–97.
Amick J, Roczniak-Ferguson A, Ferguson SM. C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol Biol Cell. 2016;27:3040–51.
Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, Halloran MA, Gleeson PA, Blair IP, Soo KY, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014;23:3579–95.
Oakes JA, Davies MC, Collins MO. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain. 2017;10:5.
Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L, Myszczynska MA, Higginbottom A, Walsh MJ, Whitworth AJ, et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016;35:1656–76.
Walker C, Herranz-Martin S, Karyka E, Liao C, Lewis K, Elsayed W, Lukashchuk V, Chiang SC, Ray S, Mulcahy PJ, et al. C9orf72 expansion disrupts ATM-mediated chromosomal break repair. Nat Neurosci. 2017;20:1225–35.
Green KM, Glineburg MR, Kearse MG, Flores BN, Linsalata AE, Fedak SJ, Goldstrohm AC, Barmada SJ, Todd PK. RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response. Nat Commun. 2017;8:2005.
Tabet R, Schaeffer L, Freyermuth F, Jambeau M, Workman M, Lee CZ, Lin CC, Jiang J, Jansen-West K, Abou-Hamdan H, et al. CUG initiation and frameshifting enable production of dipeptide repeat proteins from ALS/FTD C9ORF72 transcripts. Nat Commun. 2018;9:152.
Cheng W, Wang S, Mestre AA, Fu C, Makarem A, Xian F, Hayes LR, Lopez-Gonzalez R, Drenner K, Jiang J, et al. C9ORF72 GGGGCC repeat-associated non-AUG translation is upregulated by stress through eIF2alpha phosphorylation. Nat Commun. 2018;9:51.
Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, Kim J, Yun J, Xie Y, McKnight SL. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345:1139–45.
Yamakawa M, Ito D, Honda T, Kubo K, Noda M, Nakajima K, Suzuki N. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum Mol Genet. 2015;24:1630–45.
Mann DM, Rollinson S, Robinson A, Bennion Callister J, Thompson JC, Snowden JS, Gendron T, Petrucelli L, Masuda-Suzukake M, Hasegawa M, et al. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun. 2013;1:68.
Schludi MH, May S, Grasser FA, Rentzsch K, Kremmer E, Kupper C, Klopstock T, Arzberger T, Edbauer D. Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol. 2015;130:537–55.
Simone R, Balendra R, Moens TG, Preza E, Wilson KM, Heslegrave A, Woodling NS, Niccoli T, Gilbert-Jaramillo J, Abdelkarim S, et al. G-quadruplex-binding small molecules ameliorate C9orf72 FTD/ALS pathology in vitro and in vivo. EMBO Mol Med. 2018;10:22–31.
Su Z, Zhang Y, Gendron TF, Bauer PO, Chew J, Yang WY, Fostvedt E, Jansen-West K, Belzil VV, Desaro P, et al. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron. 2014;83:1043–50.
Cheng W, Wang S, Zhang Z, Morgens DW, Hayes LR, Lee S, Portz B, Xie Y, Nguyen BV, Haney MS, et al. CRISPR-Cas9 Screens Identify the RNA Helicase DDX3X as a Repressor of C9ORF72 (GGGGCC)n Repeat-Associated Non-AUG Translation. Neuron. 2019;104(5):885-98.e8.
Davidson YS, Barker H, Robinson AC, Thompson JC, Harris J, Troakes C, Smith B, Al-Saraj S, Shaw C, Rollinson S, et al. Brain distribution of dipeptide repeat proteins in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun. 2014;2:70.
Mackenzie IR, Frick P, Grasser FA, Gendron TF, Petrucelli L, Cashman NR, Edbauer D, Kremmer E, Prudlo J, Troost D, et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. 2015;130:845–61.
Mackenzie IR, Frick P, Neumann M. The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol. 2014;127:347–57.
Saberi S, Stauffer JE, Jiang J, Garcia SD, Taylor AE, Schulte D, Ohkubo T, Schloffman CL, Maldonado M, Baughn M, et al. Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol. 2018;135:459–74.
Sakae N, Bieniek KF, Zhang YJ, Ross K, Gendron TF, Murray ME, Rademakers R, Petrucelli L, Dickson DW. Poly-GR dipeptide repeat polymers correlate with neurodegeneration and Clinicopathological subtypes in C9ORF72-related brain disease. Acta Neuropathol Commun. 2018;6:63.
Farg MA, Konopka A, Soo KY, Ito D, Atkin JD. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum Mol Genet. 2017;26:2882–96.
Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, Monaghan J, Pandey UB, et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014;84:1213–25.
May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, Grasser FA, Mori K, Kremmer E, Banzhaf-Strathmann J, et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014;128:485–503.
Zhang YJ, Jansen-West K, Xu YF, Gendron TF, Bieniek KF, Lin WL, Sasaguri H, Caulfield T, Hubbard J, Daughrity L, et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014;128:505–24.
Boeynaems S, Bogaert E, Kovacs D, Konijnenberg A, Timmerman E, Volkov A, Guharoy M, De Decker M, Jaspers T, Ryan VH, et al. Phase Separation of C9orf72 Dipeptide Repeats Perturbs Stress Granule Dynamics. Mol Cell. 2017;65:1044–55 e1045.
Shi KY, Mori E, Nizami ZF, Lin Y, Kato M, Xiang S, Wu LC, Ding M, Yu Y, Gall JG, et al. Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc Natl Acad Sci U S A. 2017;114:E1111–7.
Brannan KW, Jin W, Huelga SC, Banks CA, Gilmore JM, Florens L, Washburn MP, Van Nostrand EL, Pratt GA, Schwinn MK, et al. SONAR Discovers RNA-Binding Proteins from Analysis of Large-Scale Protein-Protein Interactomes. Mol Cell. 2016;64:282–93.
Vanneste J, Vercruysse T, Boeynaems S, Sicart A, Van Damme P, Daelemans D, Van Den Bosch L. C9orf72-generated poly-GR and poly-PR do not directly interfere with nucleocytoplasmic transport. Sci Rep. 2019;9:15728.
Eftekharzadeh B, Daigle JG, Kapinos LE, Coyne A, Schiantarelli J, Carlomagno Y, Cook C, Miller SJ, Dujardin S, Amaral AS, et al. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer's Disease. Neuron. 2018;99:925–40 e927.
Gasset-Rosa F, Chillon-Marinas C, Goginashvili A, Atwal RS, Artates JW, Tabet R, Wheeler VC, Bang AG, Cleveland DW, Lagier-Tourenne C. Polyglutamine-Expanded Huntingtin Exacerbates Age-Related Disruption of Nuclear Integrity and Nucleocytoplasmic Transport. Neuron. 2017;94:48–57 e44.
Grima JC, Daigle JG, Arbez N, Cunningham KC, Zhang K, Ochaba J, Geater C, Morozko E, Stocksdale J, Glatzer JC, et al. Mutant Huntingtin Disrupts the Nuclear Pore Complex. Neuron. 2017;94:93–107 e106.
Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, Tatzelt J, Mann M, Winklhofer KF, Hartl FU, et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science. 2016;351:173–6.
Zhang K, Daigle JG, Cunningham KM, Coyne AN, Ruan K, Grima JC, Bowen KE, Wadhwa H, Yang P, Rigo F, et al. Stress Granule Assembly Disrupts Nucleocytoplasmic Transport. Cell. 2018a;173:958–71 e917.
Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18:285–98.
Lin Y, Mori E, Kato M, Xiang S, Wu L, Kwon I, McKnight SL. Toxic PR Poly-Dipeptides Encoded by the C9orf72 Repeat Expansion Target LC Domain Polymers. Cell. 2016;167:789–802 e712.
Gasset-Rosa F, Lu S, Yu H, Chen C, Melamed Z, Guo L, Shorter J, Da Cruz S, Cleveland DW. Cytoplasmic TDP-43 De-mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron. 2019;102:339–57 e337.
Mann JR, Gleixner AM, Mauna JC, Gomes E, DeChellis-Marks MR, Needham PG, Copley KE, Hurtle B, Portz B, Pyles NJ, et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron. 2019;102:321–38 e328.
Chen Y, Cohen TJ. Aggregation of the nucleic acid-binding protein TDP-43 occurs via distinct routes that are coordinated with stress granule formation. J Biol Chem. 2019;294:3696–706.
Neumann M, Igaz LM, Kwong LK, Nakashima-Yasuda H, Kolb SJ, Dreyfuss G, Kretzschmar HA, Trojanowski JQ, Lee VM. Absence of heterogeneous nuclear ribonucleoproteins and survival motor neuron protein in TDP-43 positive inclusions in frontotemporal lobar degeneration. Acta Neuropathol. 2007;113:543–8.
Westergard T, McAvoy K, Russell K, Wen X, Pang Y, Morris B, Pasinelli P, Trotti D, Haeusler A. Repeat-associated non-AUG translation in C9orf72-ALS/FTD is driven by neuronal excitation and stress. EMBO Mol Med. 2019;11(2):e9423.
Fay MM, Anderson PJ, Ivanov P. ALS/FTD-Associated C9ORF72 Repeat RNA Promotes Phase Transitions In Vitro and in Cells. Cell Rep. 2017;21:3573–84.
Tao Z, Wang H, Xia Q, Li K, Li K, Jiang X, Xu G, Wang G, Ying Z. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum Mol Genet. 2015;24:2426–41.
Hartmann H, Hornburg D, Czuppa M, Bader J, Michaelsen M, Farny D, Arzberger T, Mann M, Meissner F, Edbauer D. Proteomics and C9orf72 neuropathology identify ribosomes as poly-GR/PR interactors driving toxicity. Life Sci Alliance. 2018;1:e201800070.
Kanekura K, Yagi T, Cammack AJ, Mahadevan J, Kuroda M, Harms MB, Miller TM, Urano F. Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum Mol Genet. 2016;25:1803–13.
Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo EC, Krach F, Yang D, Sen A, et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell. 2018;172:590–604 e513.
Mizielinska S, Ridler CE, Balendra R, Thoeng A, Woodling NS, Grasser FA, Plagnol V, Lashley T, Partridge L, Isaacs AM. Bidirectional nucleolar dysfunction in C9orf72 frontotemporal lobar degeneration. Acta Neuropathol Commun. 2017;5:29.
White MR, Mitrea DM, Zhang P, Stanley CB, Cassidy DE, Nourse A, Phillips AH, Tolbert M, Taylor JP, Kriwacki RW. C9orf72 Poly(PR) Dipeptide Repeats Disturb Biomolecular Phase Separation and Disrupt Nucleolar Function. Mol Cell. 2019;74:713–28 e716.
Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, Narlikar GJ. Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature. 2017;547:236–40.
Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH. Phase separation drives heterochromatin domain formation. Nature. 2017;547:241–5.
Lopez-Gonzalez R, Lu Y, Gendron TF, Karydas A, Tran H, Yang D, Petrucelli L, Miller BL, Almeida S, Gao FB. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron. 2016;92:383–91.
Nihei Y, Mori K, Werner G, Arzberger T, Zhou Q, Khosravi B, Japtok J, Hermann A, Sommacal A, Weber M, et al. Poly-glycine-alanine exacerbates C9orf72 repeat expansion-mediated DNA damage via sequestration of phosphorylated ATM and loss of nuclear hnRNPA3. Acta Neuropathol. 2019.
Choi SY, Lopez-Gonzalez R, Krishnan G, Phillips HL, Li AN, Seeley WW, Yao WD, Almeida S, Gao FB. C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat Neurosci. 2019;22:851–62.
Onesto E, Colombrita C, Gumina V, Borghi MO, Dusi S, Doretti A, Fagiolari G, Invernizzi F, Moggio M, Tiranti V, et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol Commun. 2016;4:47.
Baldwin KR, Godena VK, Hewitt VL, Whitworth AJ. Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum Mol Genet. 2016;25:2378–92.
Guo L, Kim HJ, Wang H, Monaghan J, Freyermuth F, Sung JC, O'Donovan K, Fare CM, Diaz Z, Singh N, et al. Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell. 2018;173:677–92 e620.
Moore S, Alsop E, Lorenzini I, Starr A, Rabichow BE, Mendez E, Levy JL, Burciu C, Reiman R, Chew J, et al. ADAR2 mislocalization and widespread RNA editing aberrations in C9orf72-mediated ALS/FTD. Acta Neuropathol. 2019;138:49–65.
Chiriboga CA, Swoboda KJ, Darras BT, Iannaccone ST, Montes J, De Vivo DC, Norris DA, Bennett CF, Bishop KM. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology. 2016;86:890–7.
Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, Yamashita M, Rigo F, Hung G, Schneider E, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388:3017–26.
Hache M, Swoboda KJ, Sethna N, Farrow-Gillespie A, Khandji A, Xia S, Bishop KM. Intrathecal Injections in Children With Spinal Muscular Atrophy: Nusinersen Clinical Trial Experience. J Child Neurol. 2016;31:899–906.
Wahlestedt C, Salmi P, Good L, Kela J, Johnsson T, Hokfelt T, Broberger C, Porreca F, Lai J, Ren K, et al. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc Natl Acad Sci U S A. 2000;97:5633–8.
Gendron TF, Chew J, Stankowski JN, Hayes LR, Zhang YJ, Prudencio M, Carlomagno Y, Daughrity LM, Jansen-West K, Perkerson EA, et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci Transl Med. 2017;9.
Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110:E4530–9.
Sareen D, O'Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5:208ra149.
Zhou Q, Lehmer C, Michaelsen M, Mori K, Alterauge D, Baumjohann D, Schludi MH, Greiling J, Farny D, Flatley A, et al. Antibodies inhibit transmission and aggregation of C9orf72 poly-GA dipeptide repeat proteins. EMBO Mol Med. 2017;9:687–702.
Archbold HC, Jackson KL, Arora A, Weskamp K, Tank EM, Li X, Miguez R, Dayton RD, Tamir S, Klein RL, et al. TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci Rep. 2018;8:4606.
Ederle H, Funk C, Abou-Ajram C, Hutten S, Funk EBE, Kehlenbach RH, Bailer SM, Dormann D. Nuclear egress of TDP-43 and FUS occurs independently of Exportin-1/CRM1. Sci Rep. 2018;8:7084.
Pinarbasi ES, Cagatay T, Fung HYJ, Li YC, Chook YM, Thomas PJ. Active nuclear import and passive nuclear export are the primary determinants of TDP-43 localization. Sci Rep. 2018;8:7083.
Haines JD, Herbin O, de la Hera B, Vidaurre OG, Moy GA, Sun Q, Fung HY, Albrecht S, Alexandropoulos K, McCauley D, et al. Nuclear export inhibitors avert progression in preclinical models of inflammatory demyelination. Nat Neurosci. 2015;18:511–20.
Wong YL, LeBon L, Basso AM, Kohlhaas KL, Nikkel AL, Robb HM, Donnelly-Roberts DL, Prakash J, Swensen AM, Rubinstein ND, et al. eIF2B activator prevents neurological defects caused by a chronic integrated stress response. Elife. 2019;8:e42940.
Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, Messing J, Kim HJ, Soriano A, Auburger G, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature. 2017;544:367–71.
Scoles DR, Meera P, Schneider MD, Paul S, Dansithong W, Figueroa KP, Hung G, Rigo F, Bennett CF, Otis TS, et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature. 2017;544:362–6.
Fang MY, Markmiller S, Vu AQ, Javaherian A, Dowdle WE, Jolivet P, Bushway PJ, Castello NA, Baral A, Chan MY, et al. Small-Molecule Modulation of TDP-43 Recruitment to Stress Granules Prevents Persistent TDP-43 Accumulation in ALS/FTD. Neuron. 2019;103:802–19 e811.
Acknowledgements
We thank Yongjie Zhang for his critical suggestions and proofreading.
Funding
B.O. is supported by Biogen Idec, Cytokinetics, Genentech, Orion, and Eisai. K.Z. is supported by the Target ALS foundation and the Frick Foundation for ALS research.
Author information
Authors and Affiliations
Contributions
All authors wrote, read, and approved the final manuscript.
Authors’ information
Not applicable
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
B.O. is on the advisory board of MediciNova and Biogen Idec. The other authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tang, X., Toro, A., T.G., S. et al. Divergence, Convergence, and Therapeutic Implications: A Cell Biology Perspective of C9ORF72-ALS/FTD. Mol Neurodegeneration 15, 34 (2020). https://doi.org/10.1186/s13024-020-00383-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-020-00383-7