
Abstract
Background
Hereditary hemorrhagic telangiectasia (HHT) is a rare vascular disease with autosomal dominant inheritance. Disease-causing variants in endoglin (ENG) and activin A receptor type II-like 1 (ACVRL1) genes are detected in more than 90% of cases submitted to molecular diagnosis.
Methods
We used data from the RiHHTa (Computerized Registry of Hereditary Hemorrhagic Telangiectasia) registry to describe genetic variants and to assess their genotype-phenotype correlation among HHT patients in Spain.
Results
By May 2019, 215 patients were included in the RiHHTa registry with a mean age of 52.5 ± 16.5 years and 136 (63.3%) were women. Definitive HHT diagnosis defined by the Curaçao criteria were met by 172 (80%) patients. Among 113 patients with genetic test, 77 (68.1%) showed a genetic variant in ACVRL1 and 36 (31.8%) in ENG gene. The identified genetic variants in ACVRL1 and ENG genes and their clinical significance are provided. ACVRL1 mutations were more frequently nonsense (50%) while ENG mutations were more frequently, frameshift (39.1%). ENG patients were significantly younger at diagnosis (36.9 vs 45.7 years) and had pulmonary arteriovenous malformations (AVMs) (71.4% vs 24.4%) and cerebral AVMs (17.6% vs 2%) more often than patients with ACVRL1 variants. Patients with ACVRL1 variants had a higher cardiac index (2.62 vs 3.46), higher levels of hepatic functional blood tests, and anemia (28.5% vs 56.7%) more often than ENG patients.
Conclusions
ACVRL1 variants are more frequent than ENG in Spain. ACVRL1 patients developed symptomatic liver disease and anemia more often than ENG patients. Compared to ACVRL1, those with ENG variants are younger at diagnosis and show pulmonary and cerebral AVMs more frequently.
Similar content being viewed by others


Introduction
Hereditary hemorrhagic telangiectasia (HHT) or Rendu-Osler-Weber syndrome (ORPHA774; HHT 1: OMIM# 187300; HHT 2: OMIM# 600376) is a rare autosomal-dominant multisystemic vascular disease characterized by telangiectasia and larger vascular malformations (VMs), with a prevalence of 1 in 6000 [1,2,3,4]. Although more than 800 pathogenic variants in more than 5 genes have been reported, disease-causing variants in endoglin (ENG; chromosome 9q) and activin A receptor type II-like 1 (ACVRL1; chromosome 12q) genes are detected in more than 90% of cases submitted to molecular diagnosis and cause HHT1 and HHT2, respectively [5,6,7]. Mutations in SMAD4 (encoding the transcription factor Smad4) have been described in less than 2% of the HHT population and cause juvenile polyposis/HHT overlap syndrome [8]. Endoglin (encoded by ENG) is an auxiliary co-receptor at the endothelial cell surface that promotes BMP9 signaling through the activin receptor-like kinase 1 (ALK1; encoded by ACVRL1). Both proteins contribute to the signaling hub formed by BMP9-Endoglin-ALK1-Smad with a high impact in angiogenesis [9,10,11].
HHT can be diagnosed either through molecular genetic testing or using the Curaçao clinical criteria (recurrent epistaxis, cutaneous/mucosal telangiectasia, visceral involvement and a first-degree relativewith HHT) [4, 5, 7, 12]. However, since HHT is an age-dependent disease, genetic study is a very useful tool for diagnosis in young patients [4, 5, 7]. Telangiectasia is the characteristic lesion in HHT and shows dilated postcapillary venules directly connected with dilated arterioles losing the capillary bed [7, 13]. Telangiectasia in nasal mucosae causes spontaneous and recurrent epistaxis that are the most common and usually the earliest clinical manifestation of HHT [7, 12, 14]. Although endoglin and ALK1 are components of the same BMP9 receptor complex, they are structurally and functionally different proteins. Thus, pathogenic variants in their genes are related to different clinical phenotypes [5, 9]. Pulmonary arteriovenous malformations (AVMs) and brain VMs are more common in patients with HHT1, while vascular hepatic malformations are more frequent in HHT2 [4, 7, 15].
Of note, there is high inter-familial as well as intra-familial vascular involvement and clinical manifestations variability [7, 16]. Therefore, genetic study is essential for diagnostic confirmation and also for relatives’ genetic screening [5, 7, 17]. This low prevalence and clinical variability makes HHT understanding challenging, especially in uncommon situations [18]. Development of registries that include clinical and genetic data may help to overcome these difficulties, and represent a priority in the management of rare diseases [19, 20]. The use of quality registries reflects patients’ management in standard clinical practice, helps reaching a consensus on diagnostic-therapeutic measures and also generates hypotheses for developing clinical trials [19,20,21]. The aim of the present study is to describe the genetic variants and their clinical correlation in HHT patients from the RiHHTa (Computerized Registry of Hereditary Hemorrhagic Telangiectasia) registry, comprehending a cohort of individuals followed-up at specialized HHT centers in Spain [14].
Material and methods
Study design
RiHHTa is a multicenter, prospective and observational registry, developed within the Rare Diseases Working Group of the Spanish Society of Internal Medicine. RiHHTa registry is currently formed by 42 researchers from 29 Spanish hospitals. RiHHTa has an online design (accessible from https://rihhta.healthincode.com) available in Spanish or English with individual encoded access for each researcher. The rationale and methodology of RiHHTa have been published elsewhere [14]. Patients’ personal data included in the registry are in agree with Spanish personal data law (Ley 15/1999 de Protección de Datos de Carácter Personal). To safeguard the identity of the included patients, the RiHHTa registry generate an encrypted identifier for each patient. All patients (or their relatives) provided written consent for participation in the registry. The design of the RiHHTa registry was approved by the Ethics Committee of the Hospital Universitari de Bellvitge (Barcelona, Spain; ethic approval number PR241/16). The creation of RiHHTa counted on the nonremunerated collaboration of the genetic studies company Health in Code (A Coruña, Spain), which has broad experience in cardiomyopathy registries [22].
The main objective of the present study was to describe the type of genetic variants identified in HHT patients included in RiHHTa registry and to analyse the genotype-phenotype correlation according to these mutations in ENG or ACVRL1 genes.
Patient and variables
Patients included in the RiHHTa registry with a “definite” clinical diagnosis according to the Curaçao Criteria (≥ 3 criteria) or with a genetic diagnosis were considered for this study [12]. The inclusion period was from June 2016 to June 2019. Demographic data, clinical characteristics and complementary tests were collected according to mutations in ENG or ACVRL1 genes. The severity of nosebleeds was measured according to the epistaxis severity score (ESS). ESS is an on-line tool that estimates the severity of epistaxis according to clinical data occurring during the last 3 months. Epistaxis is considered moderate or severe if ESS results > 4 or > 7 points, respectively [23].
For the screening of pulmonary visceral involvement, a contrast transthoracic echocardiography (TTE) was performed to establish the degree of right-left shunt (R-L) and the need to undergo a thoracic computed tomography (CT) angiography to objectively confirm the presence of pulmonary arteriovenous malformations (AVM) [7, 24]. In addition, an abdominal CT angiography, magnetic resonance imaging (MRI) or doppler ultrasound were performed to study hepatic and/or abdominal VMs. Hepatic involvement was defined according to the three classical patterns of abnormal vascular communications: arteriovenous shunt (from hepatic artery to hepatic vein), portovenous shunt (from portal vein to hepatic vein) and arterioportal shunt (from hepatic artery to portal vein) [25]. Neurological involvement studies were carried out when neurological symptoms or family history, by a cerebral CT and/or MRI. An endoscopic digestive study was performed when there was disproportionate anaemia to the degree of epistaxis or objectively confirmed overt gastrointestinal (GI) bleeding [7].
Molecular analysis
All included patients underwent genetic testing. The sequencing technique depended on each treating centers’ preference. Because RiHHTa includes patients from all around Spain, different genetic techniques have been used in the included patients. These techniques include Next generation sequencing (NGS) technologies, Sanger sequencing and other techniques such Multiplex ligation-dependent probe amplification (MLPA). In patients in which NGS was used, coding exons and intronic boundaries of 9 genes related to HHT disease or HHT-like phenotypes (ACVRL1, BMP10, BMPR1A, BMPR2, ENG, GDF2, RASA1, SMAD1, SMAD4) were captured using a custom probe library (SureSelect Target Enrichment Kit for Illumina paired-end multiplexed sequencing method; Agilent Technologies, Santa Clara, California, USA), and sequenced on the HiSeq 1500 platform following Illumina protocols. To determine the pathogenicity of the included variants in the RiHHTa registry, all mutations were checked again by Health in Code SL (A Coruña, Spain) and labeled according to the American College of Medical Genetics and Genomics (ACMG) guidelines [26]. Whenever a genetic variant was identified through NGS, Sanger sequencing was the preferred used method for familial evaluation, unless technically not feasible.
Statistical analysis
A descriptive statistical analysis was performed for all categorical and continuous variables and expressed as proportions or means with standard deviations (SD), respectively. The descriptive analysis was performed for the entire sample. We used chi-square or Fisher’s exact test to compare categorical data between groups; Fisher’s exact test has been used when expected frequencies were less than 5. Continuous variables were compared using Student t-test. We used two-tailed unpaired t-tests to compare normally distributed continuous data between two groups, and we used the Mann-Whitney U test for non-normally distributed continuous data comparisons. Normality was assessed using Shapiro-Wilk test. P values of < 0.05 were considered statistically significant. Analyses were performed using SPSS, version 18 for the PC (SPSS, Inc. Chicago, IL, USA).
Results
Baseline characteristics
During the study period, 215 patients from 29 Spanish hospitals were included in the RiHHTa registry, 136 (63.3%) were women. Mean age at time of inclusion of 52.5 ± 16.5 years old. Most (88.4%) patients were Caucasian. The mean age at diagnosis of HHT was 42.2 ± 17.5 years old. Epistaxis was the most frequent symptom at disease onset which allowed the diagnosis of suspicion in 197 (91.6%) patients, while in another 7 (3.2%) patients the diagnosis arose after a central nervous system event (including brain abscess in 4 (1.9%) patients, and stroke, in 3 (1.4%). Family history were present in 178 (82.8%) patients and in 167 (77.7%), muco-cutaneous telangiectasia were observed at diagnosis or during the follow-up. Definite HHT diagnosis defined by the Curaçao criteria were met by 172 (80%) patients, while the remaining were diagnosed through a positive genetic test, after a thorough familial evaluation. .
Epistaxis was also the most frequent symptom at diagnosis of HHT with a mean ESS of 3.65 ± 2.5. Anemia was present in 92 (42.8%) patients, 12 (5%) patients had a history of previous venous thromboembolic disease and 8 (3.7%) patients had atrial fibrillation. Medical attention because of epistaxis at Emergency Department during follow-up was required by 69 (32.1%) patients and red blood cell transfusion by 38 (17.7%) patients.
Pulmonary AVMs were detected in 48 (22.3%) patients and liver VMs identified by imaging test (CT angiography, MRI or doppler ultrasound) were observed in 58 (27%) patients. Pancreatic involvement was detected in seven (3.3%) subjects and other abdominal VMs in 16 (7.4%) patients, including splenic, renal and gastro-omental arterial aneurysms. Brain malformations were reported in five patients (2.3%) and GI telangiectasia were observed in 26 (12.1%) patients (Table 1).
Types of pathogenic variants
A genetic test was performed in 113 HHT patients from the RiHHTa registry (Table 2). For the mutation analysis, Sanger sequencing was performed in 65 (57.5%) cases, NGS in 38 (33.6%) and other techniques, such as MLPA, in 10 (8.8%) cases. A genetic variant was identified in ACVRL1 gene in 77 (68.1%) patients, whereas 36 (31.8%) patients harboured a genetic variant in ENG gene. According to the type of mutation, ACVRL1 patients presented more frequently nonsense mutations (50.6%), while in ENG patients the most frequent type was frameshift (36.1%). Supplementary Table 1 shows those 52 variants we have complete genetic data, as well as their frequencies in gnomAD and the bioinformatic predictor’s evaluation. Supplementary Table 2 shows the ACMG classification of these variants. Among these variants, we identified 17 missense variants in ACVRL1, 41% of them were localized in the kinase domains of exons 7 and 8, and 29% in exon 3. However, missense variants in ENG gene were dispersed among the extracellular domain of the protein. In both genes, the predominant variants were the ones that caused loss-of-function mechanism (nonsense, frameshift and splice-site), which would almost certainly be pathogenic in these genes. Figure 1 shows a schematic representation of ACVRL1 and ENG, as well as the exonic variants identified.

Schematic representation of the exons (boxes) of ACVRL1 and ENG genes. The identified exonic variants in our cohort are presented according to their location (colored boxes represent specific functional domains, each one of them is explained in the legends)
Genotype-phenotype correlation
There were no significant differences between ENG and ACVRL1 patients in the age of inclusion, gender, number of Curaçao criteria, recurrent epistaxis, ESS, mucocutaneous telangiectasia, family history, hepatic VMs or GI involvement.
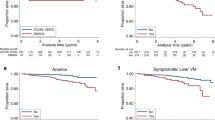
Compared to patients with ACVRL1 mutations, ENG patients were significantly younger at diagnosis (36.9 vs 45.7 years) and usually had more pulmonary AVMs (71.4 vs 24.4%), as well as cerebral AVMs (17.6% vs 2%). Those with ACVRL1 mutation had a higher cardiac index (3.46 L/min/m2 vs 2.62 L/min/m2), higher levels of hepatic functional blood tests and more frequently anemia (56.7% vs 28.5%) than ENG patients (Table 3).
Discussion
The present study using data from the RiHHTa registry represents the largest series about genetic variants of HHT patients in Spain. A previous study reported 74 clinically diagnosed Spanish patients, most of them with ACVRL1 variants [27]. In our cohort, 77 out of 113 (68.1%) patients also had variants in the ACVRL1 gene. This finding is in agreement with previous data from other Mediterranean countries but different to Northern Europe or North America, where pathogenetic variants in ENG are more frequent than in ACVRL1 [28, 29].
We have observed that patients with ENG gene variants were diagnosed at a younger age. This finding could be influenced because oral and nasal mucosal telangiectases are present earlier in life in patients with HHT1 than with HHT2, so epistaxis also start earlier in these patients [30]. According to previous studies, we have observed a higher prevalence of pulmonary AVMs and cerebral VMs in patients with ENG mutations than in those with ACVRL1 mutations (5–29%) [20, 23, 27, 31]. Hepatic involvement has been described more commonly among ACVRL1 patients, with a wide range of prevalence from 8 to 78% [29, 30, 32, 33]. In our series, though vascular hepatic involvement was more frequent in ACVRL1 patients, no statistically significant differences were detected between ENG and ACVRL1 patients, probably because different screening methods were used by RiHHTa investigators. However, patients with ACVRL1 mutation had a higher CI and levels of hepatic functional blood tests than ENG patients, suggesting a more severe hepatic involvement in patients with ACVRL1 mutations. In fact, in two studies including patients with genotype data available, those who developed symptomatic liver disease had ACVRL1 mutation [15, 34].
In our study, there were no statistically significant differences in the presence of GI telangiectasia between ENG and ACVRL1 patients. Canzonieri et al. and van Tuyl et al. systemically studied the extent of GI involvement with gastroscopy, video capsule endoscopy, and colonoscopy in 22 and 35 HHT patients, respectively, and found a higher prevalence of telangiectasia in patients with ENG mutation [8, 35, 36]. In a recent study, Mora-Luján et al. reported that age, tobacco use, ENG mutation, and hemoglobin levels were associated with GI involvement in HHT comparing 67 patients with GI disease and 28 with negative GI study [37]. However, Berg et al., Sabbà et al. and Letteboer et al. assessing genotype-phenotype relationship in HHT patients, reported a similar incidence of GI telangiectasia in HHT1 and HHT2 patients [38,39,40].
Similar to previous studies, the most frequent type of pathogenic variants we have identified in both ENG and ACVRL1 genes were those predicted to cause a loss-of-function (nonsense, frameshift and splice-site) [19, 24]. Missense variants in ACVRL1 gene were predominantly localized in the kinase domains of exons 7 and 8, while missense variants in ENG gene were dispersed in the extracellular domain of the protein [27]. In a study conducted by Sabbà et al., the correlation between phenotype and variant type was analysed in a cohort of 77 HHT2 patients. ACVRL1 patients were divided into three subgroups according to the type mutation: truncating mutations (20 patients), missense mutations (47 patients) and the c.626-3C > G splicing mutation (10 patients). Patients with c.626-3C > G mutation had a higher prevalence of pulmonary AVMs than the other two subgroups [29]. This data could be of interest for the evaluation of variants of uncertain significance and for defining useful prediction criteria based on the type of mutation. Genetic study, including the description of new pathogenic variants, usually avoid unnecessary complementary tests, especially in patients with no family history or those who meet less than 3 Curaçao criteria, as young patients [4, 5, 7, 33]. In fact, genetic study has been found preferable to the clinical Curaçao criteria among patients between 0 and 21 years [41].
Our study has some limitations and strengths that should be mentioned. First, it is an observational study from RiHHTa database. Thus, some non-measured variables could lead to a possible bias. Second, genetic and screening tests varied with local practice. Third, a variety of clinicians entered data into the registry, which may lend itself to potential inaccuracies in the data being reported. However the information obtained through RiHHTa registry is valuable and allows thorough understanding the natural history of HHT. Moreover, patients in the RiHHTa registry were from different centers in Spain, which can better reflect epidemiology data of the disease in this country.
Conclusions
In conclusion, ACVRL1 variants are most frequent than ENG variants in Spain. A description of these identified genetic variants and their clinical significance is provided. Patients with ACVRL1 variants developed symptomatic liver disease and anemia more usually than ENG patients. Compared to ACVRL1, those with ENG variants were younger at diagnosis and show pulmonary and cerebral AVMs more frequently. The RiHHTa registry can contribute to improve knowledge and management of HHT patients.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ACVRL1:
-
Activin A receptor type II-like 1
- AVMs:
-
Arteriovenous malformations
- CNS:
-
Central nervous system
- CT:
-
Computed tomography
- ENG:
-
Endoglin
- ESS:
-
Epistaxis severity score
- GI:
-
Gastrointestinal
- HHT:
-
Hereditary hemorrhagic telangiectasia
- MLPA:
-
Multiplex ligation-dependent probe amplification
- MRI:
-
Magnetic resonance imaging
- R-L:
-
Right-left
- SD:
-
Standard deviations
- TTE:
-
Transthoracic echocardiography
- VMs:
-
Vascular malformations
References
The portal for rare diseases and orphan drugs. Available online: https://www.orpha.net/consor/cgi-bin/index.php. Accessed 23 Nov 2019.
Kjeldsen AD, Vase P, Green A. Hereditary hemorrhagic telangiectasia (HHT): a population-based study of prevalence and mortality in Danish HHT patients. J Intern Med. 1999;245:31–9.
Donaldson JW, McKeever TM, Hall IP, Hubbard RB, Fogarty AW. The UK prevalence of hereditary haemorrhagic telangiectasia and its association with sex, socioeconomic status and region of residence: a population-based study. Thorax. 2014;69:161–7.
Shovlin CL, Buscarini E, Kjeldsen AD, Mager HJ, Sabba C, Droege F, et al. European reference network for rare vascular diseases (VASCERN) outcome measures for hereditary Haemorrhagic telangiectasia (HHT). Orphanet J Rare Dis. 2018;13:136.
McDonald J, Wooderchak-Donahue W, VanSant WC, Whitehead K, Stevenson DA, Bayrak-Toydemir P. Hereditary hemorrhagic telangiectasia: genetics and molecular diagnostics in a new era. Front Genet. 2015;6:1.
Prigoda NL, Savas S, Abdalla SA, Piovesan B, Rushlow D, Vandezande K, et al. Hereditary haemorrhagic telangiectasia: mutation detection, test sensitivity and novel mutations. J Med Genet. 2006;43:722–8.
Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, et al. HHT Foundation International - Guidelines Working Group International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011;48:73–87.
Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet. 2004;363:852–9.
Tillet E, Bailly S. Emerging roles of BMP9 and BMP10 in hereditary hemorrhagic telangiectasia. Front Genet. 2015;5:456.
Alsina-Sanchís E, García-Ibáñez Y, Figueiredo AM, Riera-Domingo C, Figueras A, Matias-Guiu X, et al. ALK1 loss results in vascular hyperplasia in mice and humans through PI3K activation. Arterioscler Thromb Vasc Biol. 2018;38:1216–29.
Iriarte A, Figueras A, Cerdà P, Mora JM, Jucglà A, Penín R, et al. PI3K (Phosphatidylinositol 3-Kinase) Activation and Endothelial Cell Proliferation in Patients with Hemorrhagic Hereditary Telangiectasia Type 1. Cells. 2019;8(9):971.
Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu- Osler-weber syndrome). Am J Med Genet. 2000;91:66–7.
Braverman IM, Keh A, Jacobson BS. Ultrastructure and three-dimensional organization of the telangiectases of hereditary hemorrhagic telangiectasia. J Invest Dermatol. 1990;95:422–7.
Riera-Mestre A, Mora Luján JM, Sanchez Martínez R, Torralba Cabeza MA, de la Peña JL P, Juyol Rodrigo MC, et al. Computerized registry of patients with hemorrhagic hereditary telangiectasia (RiHHTa Registry) in Spain: Objectives, methods, and preliminary results. Rev Clin Esp. 2018;218:468–76.
Lesca G, Olivieri C, Burnichon N, Pagella F, Carette M-F, Gilbert-Dussardier B, et al. Genotype-phenotype correlations in hereditary hemorrhagic telangiectasia: data from the French-Italian HHT network. Genet Med. 2007;9:14–22.
McDonald J, Bayrak-Toydemir P, Pyeritz RE. Hereditary hemorrhagic telangiectasia: an overview of diagnosis, management and pathogenesis. Genet Med. 2011;13:607–16.
Van Gent MW, Velthuis S, Post MC, Snijder RJ, Westermann CJ, Letteboer TG, et al. Hereditary hemorrhagic telangiectasia: how accurate are the clinical criteria? Am J Med Genet A. 2013;161A:461–6.
Riera-Mestre A, Mora-Luján JM, Trujillo-Santos J, Del Toro J, Nieto JA, Pedrajas JM, RIETE Investigators, et al. Natural history of patients with venous thromboembolism and hereditary hemorrhagic telangiectasia. Findings from the RIETE registry. Orphanet J Rare Dis. 2019;14:196.
D’Agnolo HM, Kievit W, Andrade RJ, Karlsen TH, Wedemeyer H, Drenth JP. Creating an effective clinical registry for rare diseases. Unit Eur Gastroenterol J. 2016;4:333–8.
EURORDIS-NORD-CORD Joint Declaration of 10 Key Principles for Rare Disease Patient Registries. Available from: http://download.eurordis.org/documents/pdf/EURORDIS_NORD_CORD_JointDec_Registries_FINAL.pdf. Accessed 15 Sept 2019.
Kodra Y, Posada de la Paz M, Coi A, Santoro M, Bianchi F, Ahmed F, et al. Data quality in rare diseases registries. Adv Exp Med Biol. 2017;1031:149–64.
Torrado M, Maneiro E, Trujillo-Quintero JP, Evangelista A, Mikhailov AT, Monserrat L. A novel heterozygous Intronic mutation in the FBN1 gene contributes to FBN1 RNA Missplicing events in the Marfan syndrome. Biomed Res Int. 2018;2018:3536495.
Hoag JB, Terry P, Mitchell S, Reh D, Merlo CA. An epistaxis severity score for hereditary hemorrhagic telangiectasia. Laryngoscope. 2010;120:838–43.
Barzilai B, Waggoner AD, Spessert C, Picus D, Goodenberger D. Two-dimensional contrast echocardiography in the detection and follow-up of congenital pulmonary arteriovenous malformations. Am J Cardiol. 1991;68:1507–10.
Khalid SK, Garcia-Tsao G. Hepatic vascular malformations in hereditary hemorrhagic telangiectasia. Semin Liver Dis. 2008;28:247–58.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG laboratory quality assurance committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association of Molecular Pathology. Genet Med. 2015;17:405–24.
Fontalba A, Fernandez-L A, García-Alegria E, Albiñana V, Garrido-Martin EM, Blanco FJ, et al. Mutation study of Spanish patients with hereditary hemorrhagic telangiectasia. BMC Med Genet. 2008;9:75.
Olivieri C, Pagella F, Semino L, Lanzarini L, Valacca C, Pilotto A, et al. Analysis of ENG and ACVRL1 genes in 137 HHT Italian families identifies 76 different mutations (24 novel). Comparison with other European studies. J Hum Genet. 2007;52:820–9.
Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–95.
Letteboer TGW, Mager H-J, Snijder RJ, Lindhout D, Ploos van Amstel H-K, Zanen P, Westermann KJJ. Genotype–phenotype relationship for localization and age distribution of telangiectases in hereditary hemorrhagic telangiectasia. Am J Med Genet. 2008;146A:2733–9.
Bayrak-Toydemir P, McDonald J, Markewitz B, Lewin S, Miller F, Chou LS, et al. Genotype-phenotype correlation in hereditary hemorrhagic telangiectasia: mutations and manifestations. Am J Med Genet A. 2006;140:463–70.
Buscarini E, Gandolfi S, Alicante S, Londoni C, Manfredi G. Liver involvement in hereditary hemorrhagic telangiectasia. Abdom Radiol (NY). 2018;43:1920–30.
Riera-Mestre A, Ribas J, Castellote J. Medical management of haemorrhagic hereditary telangiectasia in adult patients. Med Clin (Barc). 2019;152:274–80.
Singh S, Swanson KL, Hathcock MA, Kremers WK, Pallanch JF, Krowka MJ, et al. Identifying the presence of clinically significant hepatic involvement in hereditary haemorrhagic telangiectasia using a simple clinical scoring index. J Hepatol. 2014;61:124–31.
Canzonieri C, Centenara L, Ornati F, Pagella F, Matti E, Alvisi C, et al. Endoscopic evaluation of gastrointestinal tract in patients with hereditary hemorrhagic telangiectasia and correlation with their genotypes. Genet Med. 2014;16:3–10.
Van Tuyl SA, Letteboer TG, Rogge-Wolf C, Kuipers EJ, Snijder RJ, Westermann CJ, et al. Assessment of intestinal vascular malformations in patients with hereditary hemorrhagic teleangiectasia and anemia. Eur J Gastroenterol Hepatol. 2007;19:153–8.
Mora-Luján JM, Iriarte A, Alba E, Sánchez-Corral MA, Berrozpe A, Cerdà P, et al. Gastrointestinal Bleeding in Patients with Hereditary Hemorrhagic Telangiectasia: Risk Factors and Endoscopic Findings. J Clin Med. 2019;9(1):82.
Berg J, Porteous M, Reinhardt D, Gallione C, Holloway S, Umasunthar T, et al. Hereditary haemorrhagic telangiectasia: a questionnaire based study to delineate the different phenotypes caused by endoglin and ALK1 mutations. J Med Genet. 2003;40:585–90.
Sabbà C, Pasculli G, Lenato GM, Suppressa P, Lastella P, Memeo M, et al. Hereditary hemorrhagic telangiectasia: clinical features in ENG and ALK1 mutation carriers. J Thromb Haemost. 2007;5:1149–57.
Letteboer TG, Mager JJ, Snijder RJ, Koeleman BP, Lindhout D, Ploos van Amstel JK, et al. Genotype-phenotype relationship in hereditary haemorrhagic telangiectasia. J Med Genet. 2006;43:371–7.
Pahl KS, Choudhury A, Wusik K, Hammill A, White A, Henderson K, et al. Applicability of the Curaçao criteria for the diagnosis of hereditary hemorrhagic telangiectasia in the pediatric population. J Pediatr. 2018;197:207–3.
Acknowledgements
We express our gratitude to Health in Code (A Coruña, Spain) for supporting this Registry. We also thank the patients enrolled in RiHHTa registry.
Coordinator of the RiHHTa Registry: Riera-Mestre A.
Members of the RiHHTa Registry of theRare Diseases Working Groupfrom theSpanish Society of Internal Medicine:
Alonso-Cotoner C, Añón S, Beneyto M, MM Bermejo-Olano, Cerdà P, Cruellas F, De Los Santos A, Díez L, Fernández A, García-Morillo JS, Gil R, Gómez-Cerezo JF, Gómez del Olmo V, González-García A,Iriarte A, Iglesias P, Juyol MC, López-Osle N, López M, López-Wolf D, Mora-Luján JM, Moreno M, Ojeda A, Patier JL, Pérez de León JA, Perez ML, Riera-Mestre A, Rivera S, Rodríguez S, Sánchez-Martínez R, Torralba MA, Zarrabeitia R.
Funding
No funding was provided for this study.
Author information
Authors and Affiliations
Consortia
Contributions
RSM, AI and ARM conceptualized and designed the study; all authors acquired the data; RSM, AI, JSM and ARM analysed the data and interpreted the findings; AI and ARM performed statistical analysis; JSM reviewed genetic data; all authors contributed to the draft of the manuscript, read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Clinical Research Ethics Committee of the Hospital Universitari de Bellvitge (Barcelona, Spain; ethic approval number PR241/16).
Consent for publication
This manuscript does not contain any individual person’s data in any form. Each patient was identified by a unique alphanumeric identification code and all data were made anonymous and analysed as aggregates.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Table S1. Identified genetic variants in ACVRL1 and ENG genes. Table S2. Clinical significance of the identified variants according to ACMG criteria.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sánchez-Martínez, R., Iriarte, A., Mora-Luján, J.M. et al. Current HHT genetic overview in Spain and its phenotypic correlation: data from RiHHTa registry. Orphanet J Rare Dis 15, 138 (2020). https://doi.org/10.1186/s13023-020-01422-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-020-01422-8