
Abstract
Background
DNA Ligase IV (LIG4) syndrome is a rare disease with few reports to date. Patients suffer from a broad spectrum of clinical features, including microcephaly, growth retardation, developmental delay, dysmorphic facial features, combined immunodeficiency, and malignancy predisposition. There may be a potential association between genotypes and phenotypes. We investigated the characteristics of LIG4 syndrome in a Chinese cohort.
Results
All seven patients had growth restriction. Most patients (6/7) had significant microcephaly (< − 3 SD). Recurrent bacterial infections of the lungs and intestines were the most common symptoms. One patient had myelodysplastic syndromes. One patient presented with an inflammatory bowel disease (IBD)-like phenotype. Patients presented with combined immunodeficiency. The proportions of naïve CD4+ and naïve CD8+ T cells decreased notably in five patients. All patients harbored compound heterozygous mutations in the LIG4 gene, which consisted of a missense mutation (c.833G > T, p.R278L) and a deletion shift mutation, primarily c.1271_1275delAAAGA (p.K424Rfs*20). Two other deletion mutations, c.1144_1145delCT and c.1277_1278delAA, were novel. Patients with p.K424Rfs*20/p.R278 may have milder dysmorphism but more significant IgA/IgM deficiency compared to the frequently reported genotype p.R814X/p.K424Rfs*20. One patient underwent umbilical cord blood stem cell transplantation (UCBSCT) but died.
Conclusions
The present study reported the clinical and molecular characteristics of a Chinese cohort with LIG4 syndrome, and the results further expand the phenotypic and genotypic spectrum and our understanding of genotype-to-phenotype correlations in LIG4 syndrome.
Similar content being viewed by others


Background
DNA ligase IV (LIG4) syndrome is an exceptionally rare autosomal recessive disorder that belongs to the group of hereditary diseases associated with impaired DNA damage-response mechanisms. The distinct features of LIG4 syndrome patients are microcephaly, growth retardation, developmental delay, dysmorphic facial features, variable immunodeficiency, pancytopenia, malignancy predisposition and pronounced clinical and cellular radiosensitivity [1].
DNA ligase IV functions in the nonhomologous end-joining (NHEJ) pathway, which is a major mechanism involved in the repair of DNA double-strand breaks (DSBs) in mammals [2], and it leads to an increased sensitivity to ionizing radiation. DSBs are also involved in the processes of class switch and V(D)J recombination during immune development. Therefore, failure to perform the processes caused by NHEJ deficiency confers (severe) combined immunodeficiency.
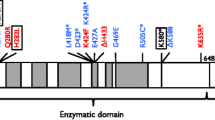
The LIG4 gene maps to chromosome 13q33-q34, and it has a complex structure formed by four domains: the DNA-binding domain (DBD), adenylation domain (AdD), oligo-binding domain (OBD) and XRCC4-binding domain (XBD). LIG4 syndrome is caused by homozygous or compound heterozygous mutations in the LIG4 gene, and the most common genotype is p.R814X/p.K424Rfs*20 [3]. Murray et al. [4] first proposed a genotype–phenotype correlation between the position of truncating mutations associated with p.R814X and disease severity.
Although this disease was first described nearly 30 years ago, only a few cases have been reported to date [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27]. The present study reported the clinical, immunological and genetic characteristics of 7 Chinese patients with LIG4 syndrome.
Methods
The Ethics Committee of the Children’s Hospital of Fudan University approved this study. Informed consent was obtained from the parents of the patients.
Patients and clinical data
LIG4 syndrome was suspected based on the clinical manifestations of patients referred to our hospital between July 2014 and December 2019 and was further confirmed via immune function and gene detection in the present study. The relevant data are summarized in detail. Previous patients and mutations reported in PubMed Medline (https://www.ncbi.nlm.nih.gov/pubmed/) were reviewed and compared.
Immunological function
Routine blood counts and immunological function analyses were performed. We used nephelometry to detect immunoglobulins, including IgG, IgA, and IgM, and lymphocyte subsets were measured using flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA). The following validated antibodies were used for flow cytometry: anti-CD3 (UCHT1), anti-CD8 (RPAT8), anti-CD27 (M-T271), anti-CD45RA (HI100), anti-CD4 (RPA-T4), anti-TCRαβ (T10B9.1A-31), anti-TCRγδ (B1), anti-CD19 (HIB19), anti-CD24 (ML5), anti-CD38 (HIT2), and anti-IgD (IA6–2) (all from BD Biosciences).
Molecular analysis
Genomic DNA was extracted from the peripheral blood of the patients and their parents using the QIAamp DNA Blood Mini kit (Qiagen, Hilden, Germany). DNA quality was assessed using a NanoDrop ultraviolet spectrophotometer (Thermo Fisher Scientific, USA).
Next-generation sequencing was performed using a panel that included all previously reported immunodeficiency genes. Genomic DNA fragments of patients were ligated with adaptors so that two paired-end DNA libraries with insert sizes of 500 bp were formed for all samples. The DNA libraries after enrichment were sequenced on the HiSeq 2000 platform in accordance with the manufacturer’s instructions (Illumina, San Diego, CA). The variants were annotated in ANNOVAR and VEP software and predicted with SIFT, PolyPhen-2 and MutationTaster. The mutations were confirmed using Sanger sequencing.
Results
Clinical manifestations
Overview
Seven patients (4 males and 3 females) were diagnosed over a 5-year period in our center. The average age of morbidity was 5.3 months (range, 1 week-14 months), and the mean time of diagnosis was extended to 18.4 months. All of these cases were full-term infants. No patients were born out of consanguineous marriages. No disease-related family histories were found, except the mother of patient 2 (P2) and P5 had a previous pregnancy with embryo growth arrest. The clinical findings are summarized in Table 1.
Microcephaly and growth restriction was obvious in these patients. The head circumference of six patients was more than 3 SD below the population mean of the same age and gender. One patient had a head circumference between 2 and 3 SD below the mean. Most of the patients had postnatal underweight (6 of 7 < − 3 SD) and stature lower than standard (2 of 7 < − 3 SD), and the weight loss was more pronounced (Fig. 1). Three full-term, small-for-gestational-age infants indicated intrauterine growth retardation during the early life stage. Two patients had clinical developmental retardation, which was primarily manifested as delayed language and motor retardation compared to healthy children of the same age. Head MRI of five patients showed no abnormalities. Intelligence assessment of scales or questionnaires was only completed in P1 and P2. However, the typical facial deformity was not found in our study.

Growth index of patients with LIG4 syndrome compared to healthy children in China. Height is marked in blue, and weight is marked in red. M: male. F: female
Infection characteristics
Within a few months after birth, these patients developed conditions like pneumonia, diarrhea, thrush, and otitis media, and one patient presented with omphalitis shortly in the early stage. Acinetobacter, Enterobacter cloacae, Flavobacterium menigosepticum and Haemophilus influenzae were generally detected from the secretion of lower respiratory tracts via bacterial culture. Salmonella was cultured in the stool of two patients with diarrhea. Parainfluenza virus was also detected repeatedly from sputum in P1 and P4. Despite oral antiviral and interferon inhalation therapy, the virus was difficult to clear. Pulmonary CT of P1 showed diffuse interstitial changes (Fig. 2), which highly correlated with viral infection. Metagenomic sequencing of the bronchoalveolar lavage fluid sample in P4 detected Pneumocystis jiroveci (reads, 189), which partially caused the extensive lung involvement.

Clinical data of patients with LIG4 syndrome. a: Facial features of P7. b: The axial (1) and coronal (2) CT scan of the chest (P1) showed bilateral lung diffuse lesions with opacification. c: Gastroenterological endoscope examination of P2 demonstrated multiple ulcers of the oropharynx (1), small intestine (2), colon (3) and rectum (4). d: Morphological examination of bone marrow (P6) revealed abnormal hematopoiesis in granulocyte and erythroid series to different extents. Vacuolar degeneration (1), binucleated (2) or giant (3) granulocytes were observed in granulocyte series. Erythroid series were active proliferation, megaloblastic change and occasionally positive PAS staining (4)
Autoimmune and other characteristics
Patients may also present with noninfectious manifestations. Routine blood analysis revealed low platelet counts in 3 patients and increased counts after intravenous immune globulin (IVIG) infusion. Other blood system abnormalities, such as leukopenia, occurred in two children. A laboratory examination of P6 suggested pancytopenia at 26 months, and she was suspected of having “myelodysplastic syndromes” based on a bone marrow aspiration smear. However, a bone marrow biopsy was not performed further. Three cases had skin lesions, and one of these patients (P4) presented with erythroderma and one patient with hypopigmentation. P5 primarily suffered from recurrent eczema and skin peeling, accompanied by an increase of eosinophilic cell count to 2.7–8.1 × 109/L.
Notably, P2 was initially admitted to our hospital due to oral ulcers and intermittent fever. Therapy with multiple antibiotics, such as mepin, vancomycin, SMZ and itraconazole, was ineffective, and only respiratory syncytial virus in sputum was detected as positive. Colonoscopy revealed multiple ulcers (Fig. 2), but no etiology was found in biopsy samples using metagenomic sequencing. After steroid administration, the body temperature was lower, and the oral ulcers were slightly better.
Immune function evaluation
Flow cytometric analysis of peripheral blood showed a significant decrease in the absolute numbers of CD19+ B cells and CD3+ T cells (especially CD4+ T cells) but normal or decreased numbers of NK cells (Additional file 1: Table S1). However, the lymphocyte count was decreased slightly in P5, in whom a chimerism test was not performed to exclude maternal-fetal cellular trafficking. Peripheral blood lymphocyte subsets of five patients (P1, P2, P3, P6, and P7) were analyzed in detail. All of these cases exhibited significantly decreased naïve Th cells (CD4 + CD45RA + CD27+) and naïve cytotoxic T cells (CD8 + CD45RA + CD27+) compared to the reference values in healthy children in China [28]. The IgG levels of P5 and P7 were significantly lower than the normal reference value and accompanied by reduced IgM. Other patients had normal levels of IgG, but three patients were tested after IVIG infusion. Comparisons of immune function between this study and previous reports [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27] are shown in Fig. 3.

Comparison of clinical presentation and immunophenotyping between this cohort and ~ 55 previously reported patients. Children whose IgG levels were detected after IVIG infusion were not included
Genetic characteristics
Mutations in the LIG4 gene were identified in all 7 cases. Patients harbored compound heterozygous mutations that consisted of a missense mutation and a deletion shift mutation. The common missense mutation c.833G > T in exon 2, which causes an amino acid change of Arg to Leu, was identified. In addition, c.1271_1275delAAAGA was found in four patients and became the most frequent deletion mutation. Three other deletion mutations were also detected, specifically c.1144_1145delCT, c.1277_1278delAA and c.1270_1274delAAAAG (Table 2). Mutations c.1144_1145delCT and c.1277_1278delAA were not reported previously and are predicted to be deleterious by Mutation Taster (value 1).
The most frequent genotype in our cohort was p.R278L/p.K424Rfs*20, which was found in five children. Further comparison with p.R814X/p.K424Rfs*20 reported in previous studies [4, 23] revealed a small difference in phenotype (Additional file 2: Table S2). Facial dysmorphism were more common in patients with genotype p.R814X/p.K424Rfs*20 (7/7) than genotype p.R278/p.K424Rfs*20 (0/9) [3, 19, 24]. Patients with p.R814X/p.K424Rfs*20 had primarily IgG deficiencies, and patients with p.R278/p.K424Rfs*20 were accompanied by a decrease in IgA and IgM levels.
Treatment and outcome
These patients underwent antibiotic and symptomatic treatment. The choice of empirical antibiotics was primarily based on the immune and infection characteristics and included bacteria, fungi and pneumocystis. Intravenous immunoglobulin was infused as supportive treatments depending on the response. However, most patients (5/7) died or gave up on treatment without transplantation due to severe infection at early stages of the disease. Only P1 received an HLA-matched unrelated HSCT using umbilical cord blood at 29 months. At the time of HSCT, the child had severe pulmonary infection and watery diarrhea with stools 4–6 times per day. Fludarabine and Busulfex were initiated as the conditioning regimen, and cyclosporine A (CsA) was used as GvHD prophylaxis. After transplantation, the child suffered from recurrent fever and aggravated cough. Flavobacterium menigosepticum and Parainfluenza virus 3 were detected in the sputum. Despite supportive treatments with multiple antibiotics and immunoglobulin replacement therapy, this child became worse. Unfortunately, he died half a month later because of respiration-circulation failure and severe inflammation with unsuccessful engraftment (chimerism 0.11% at 7 days).
Discussion
DNA ligase IV deficiency is a rare primary immunodeficiency that was first reported by Plowman in 1990 [29]. Since then, 55 patients (44 non-Chinese and 11 Chinese) were reported in PubMed Medline with a broad spectrum of clinical features [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27]. We described the clinical and laboratory features of 7 patients with LIG4 mutations in a Chinese cohort.
Our study found no obvious family history, except for P2 and P5. Both mothers had abnormal pregnancies with embryo growth arrest. DNA ligase IV is essential for embryonic viability, and knockout mice died at an early embryonic stage [30]. The six patients in this cohort presented with significant microcephaly, and all seven patients had growth restriction, which confirms that these two manifestations are the most prominent features of LIG4 syndrome, as reported previously. Therefore, LIG4 syndrome may be considered when patients present with repeated infection and a head circumference more than 3 SD below the population mean. Developmental delay also occurred in two of our patients. The impairment in expressive language skills was predominant in LIG4 patients [4]. It is necessary to perform early intelligence assessment on patients with microcephaly, even if head MRI shows no apparent abnormality.
Although facial dysmorphism, such as “bird-like” or “Seckel syndrome-like”, are always observed in LIG4 patients, none of our patients had these descriptions in their medical history. Other symptoms, including thrombocytopenia, leukopenia, pancytopenia, and skin lesions, were observed in our patients, except for bone abnormalities. Three patients developed thrombocytopenia. An elevated number of platelets after IVIG infusion suggested potential autoimmune factors, although the results of a routine autoantibody test was negative. One patient manifested as pancytopenia at the age of 2 years, and this patient was suspected of having “myelodysplastic syndromes” (MDS) based on a bone marrow aspiration smear. Most of the patients were at a very young age, so thrombocytopenia may have been more evident than cytopenia. Pancytopenia may mark progression to bone marrow failure, and MDS was reported previously [12]. The pathogeny of MDS was not thoroughly elucidated, but reduced telomere length was found in LIG4 syndrome, which is analogous to other genetically unstable diseases, such as Fanconi anemia and dyskeratosis congenita. We suspected that the accumulation of DSBs in myeloid progenitors led by environmental or infectious triggers and/or autoimmune disorders, may also contribute to bone marrow failure.
Notably, P2 presented with recurrent fever, and colonoscopy indicated extensive intestinal ulceration. To the best of our knowledge, this report is the first patient with an IBD-like phenotype. In previously reported cases, only one patient with LIG4 syndrome was documented to present a phenotype that mimicked Bechet’s disease (BD) [26], and one female patient who was diagnosed with IBD harbored a heterozygous stop-gain (p.R814X) variant in the LIG4 gene [31]. IBD-like immunopathology is a common finding in patients with complex defects in T- and B-cell function, such as Wiskott Aldrich syndrome (WAS) and atypical SCID [32] or Omenn syndrome. Although no further information was found, it seems reasonable to assume the LIG4 gene as a candidate gene for IBD [33]. An abnormal immune reaction in LIG4 patients is likely a contributor. Susceptibility to malignancy is common in disorders influencing double-strand DNA break damage repair. Eleven patients were reported [3, 5,6,7, 9, 11, 15, 18, 19, 26] whose median onset age of malignancy was 4 years old. However, no malignancy occurred in our study, likely because of the early death of these patients.
NHEJ is also important for immune development. Therefore, LIG4 patients manifest as (severe) combined immunodeficiency. Among the reported cases, approximately three quarters of patients suffered from recurrent infection with varying degrees of severity. In our case series, recurrent pneumonia and diarrhea were the most common symptoms. Pathogens, including Streptococcus pneumoniae, Haemophilus influenzae, CMV, Candida albicans, Salmonella typhimurium, and EBV, are frequently detected in LIG4 patients. Exhibiting a slight difference, the Acinetobacter, Parainfluenza virus and Salmonella infections are also prominent. The immunological detection showed a profound T (especially CD4+ T) and B lymphocytopenia with hypogammaglobulinemia in most of our patients. One patient just showed slightly decreased lymphocytes, and we suspect that this may reflect compensatory mechanisms of the proliferation of T cells or chimerism, such as maternal-fetal cellular trafficking. Further analysis of lymphocyte subsets in our patients revealed that the proportion of naïve CD4+ and naïve CD8+ T cells were markedly decreased, and memory T cells were increased, which is consistent with the three siblings reported by Felgentreff et al. [21]. The decreased naïve T cells may lead to ineffective resistance to primary infection. All patients in this cohort had significantly reduced B cell counts, but some patients had normal immunoglobulin levels, likely due to the ability of the remaining B cells to develop into plasma cells and produce antibodies.
Nearly 85% of reported patients carry compound heterozygous mutations in the LIG4 gene, and the most frequent alleles are R814X and K424fs in non-Chinese cases [3]. Mutation K424fs causes a premature stop codon 20 aa downstream, and the protein expression level in skin fibroblasts is notably reduced [8]. Truncating mutation R814X, which is at the XRCC4 binding domain, retained 10% ~ 15% residual double-strand ligation activity [6]. However, the slight difference is that two mutations R278L and K424fs account for most Chinese patients [19, 25]. The R278L mutation resides in the vicinity of the ATP-binding site. We predict that the function mechanism may be similar to the previously reported R278H mutation at the same position. Interaction between the ligase and XRCC4 is not affected, but the enzyme–adenylate complex formation is severely impaired, which results in the ~ 10% residual ligase activity [34, 35]. In all LIG4-mutated patients, the most prevalent genotypes are p.R814X associated with another truncating mutation, especially p.R814X/p.K424Rfs*20, and p.K424Rfs*20 associated with mutations near active site (K273) is another frequent genotype [23]. The phenotypes of these two genotypes are also slightly different from each other. Notably, p.K424Rfs*20/p.R278L is the only genotype seen in the Chinese population [19], and two patients with p.K424Rfs*20/p.R278H were reported in other countries [3, 24], which highlights the significance of the genotype p.K424Rfs*20/p.R278. Further comparison with the genotype p.R814X/p.K424Rfs*20 [4, 23], suggests that p.K424Rfs*20/p.R278 leads to milder dysmorphism, but more significant IgA/IgM deficiency. Hypomorphic mutations in ligase IV, such as R278H, may allow normal development but confer marked radiosensitivity, as reported in previous studies [35]. Decreased IgA/IgM levels may be associated with early clinical digestive symptoms, similar to the disease selective IgA deficiency. Patients with p.K424Rfs*20/p.R278 are at higher risk of severe infections. These findings deepen our understanding of genotype-to-phenotype correlations.
The treatment of LIG4 syndrome primarily includes antibiotic prophylaxis, immunoglobulin substitution, transfusion support and avoiding unnecessary exposure to ionizing radiation [1]. Hematopoietic stem cell transplantation (HSCT) has been performed in 10 patients [8, 9, 12,13,14, 17, 22, 23, 25], six of whom were successful, but the others died because of different complications, primarily infections. Notably, microcephaly or neurodevelopmental delay in these patients cannot be cured with HSCT. Hematopoietic stem cell transplantation requires cytotoxic agents, especially CsA [36], which may induce DSBs and be harmful for patients with this syndrome. Therefore, personalized transplant conditioning should be performed carefully in these patients. Only one patient in our study underwent HSCT and died due to severe infection. Other patients received supportive treatment. Notably, P2 was treated with glucocorticoids due to persistent fever and IBD. Immunosuppressant was also used in the reported patient who presented with a BD-like phenotype.
Conclusions
In summary, we reported the clinical manifestations and treatment of seven Chinese patients with LIG4 syndrome. The finding of the IBD-like phenotype may expand the phenotypic spectrum of this disease. R278L and K424fs were two common mutations, and p.K424Rfs*20/p.R278L was the only genotype seen in the Chinese population. Genotype p.K424Rfs*20/p.R278 might lead to milder dysmorphism but more significant IgA/IgM deficiency compared to p.R814X/p.K424Rfs*20. Genotype-to-phenotype correlations were further understood.
Availability of data and materials
The datasets used and/or analysed during the current study are all included within the article and available from the corresponding author on reasonable request.
Abbreviations
- BD:
-
Behçet’s disease
- DSBs:
-
DNA double strand breaks
- GvHD:
-
Graft-versus-host disease
- HLA:
-
Human leukocyte antigen
- HSCT:
-
Haematopoietic stem cell transplant
- IBD:
-
Inflammatory bowel disease
- IVIG:
-
Intravenous immune globulin
- LIG4:
-
DNA Ligase IV
- MDS:
-
Myelodysplastic syndromes
- NGS:
-
Next-generation sequencing
- NHEJ:
-
Nonhomologous end-joining
- UCBSCT:
-
Umbilical cord blood stem cell transplantation
References
Altmann T, Gennery AR. DNA ligase IV syndrome; a review. Orphanet J Rare Dis. 2016;11(1):137.
Helleday T, Lo J, van Gent DC, Engelward BP. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst). 2007;6(7):923–35.
Staines BA, Chinn IK, Alaez-Verson C, Yamazaki-Nakashimada MA, Carrillo-Sanchez K, Garcia-Cruz M, et al. Failing to make ends meet: the broad clinical Spectrum of DNA ligase IV deficiency. Case series and review of the literature. Front Pediatr. 2018;6:426.
Murray JE, Bicknell LS, Yigit G, Duker AL, van Kogelenberg M, Haghayegh S, et al. Extreme growth failure is a common presentation of ligase IV deficiency. Hum Mutat. 2014;35(1):76–85.
Riballo E, Critchlow SE, Teo SH, Doherty AJ, Priestley A, Broughton B, et al. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr Biol. 1999;9(13):699–702.
O'Driscoll M, Cerosaletti KM, Girard PM, Dai Y, Stumm M, Kysela B, et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol Cell. 2001;8(6):1175–85.
Ben-Omran TI, Cerosaletti K, Concannon P, Weitzman S, Nezarati MM. A patient with mutations in DNA ligase IV: clinical features and overlap with Nijmegen breakage syndrome. Am J Med Genet A. 2005;137A(3):283–7.
Buck D, Moshous D, de Chasseval R, Ma Y, le Deist F, Cavazzana-Calvo M, et al. Severe combined immunodeficiency and microcephaly in siblings with hypomorphic mutations in DNA ligase IV. Eur J Immunol. 2006;36(1):224–35.
Enders A, Fisch P, Schwarz K, Duffner U, Pannicke U, Nikolopoulos E, et al. A severe form of human combined immunodeficiency due to mutations in DNA ligase IV. J Immunol. 2006;176(8):5060–8.
van der Burg M, van Veelen LR, Verkaik NS, Wiegant WW, Hartwig NG, Barendregt BH, et al. A new type of radiosensitive T-B-NK+ severe combined immunodeficiency caused by a LIG4 mutation. J Clin Invest. 2006;116(1):137–45.
Toita N, Hatano N, Ono S, Yamada M, Kobayashi R, Kobayashi I, et al. Epstein-Barr virus-associated B-cell lymphoma in a patient with DNA ligase IV (LIG4) syndrome. Am J Med Genet A. 2007;143A(7):742–5.
Gruhn B, Seidel J, Zintl F, Varon R, Tonnies H, Neitzel H, et al. Successful bone marrow transplantation in a patient with DNA ligase IV deficiency and bone marrow failure. Orphanet J Rare Dis. 2007;2:5.
Grunebaum E, Bates A, Roifman CM. Omenn syndrome is associated with mutations in DNA ligase IV. J Allergy Clin Immunol. 2008;122(6):1219–20.
Unal S, Cerosaletti K, Uckan-Cetinkaya D, Cetin M, Gumruk F. A novel mutation in a family with DNA ligase IV deficiency syndrome. Pediatr Blood Cancer. 2009;53(3):482–4.
Yue J, Lu H, Lan S, Liu J, Stein MN, Haffty BG, et al. Identification of the DNA repair defects in a case of Dubowitz syndrome. PLoS One. 2013;8(1):e54389.
IJspeert H, Warris A, van der Flier M, Reisli I, Keles S, Chishimba S, et al. Clinical spectrum of LIG4 deficiency is broadened with severe dysmaturity, primordial dwarfism, and neurological abnormalities. Hum Mutat. 2013;34(12):1611–4.
Tamura S, Higuchi K, Tamaki M, Inoue C, Awazawa R, Mitsuki N, et al. Novel compound heterozygous DNA ligase IV mutations in an adolescent with a slowly-progressing radiosensitive-severe combined immunodeficiency. Clin Immunol. 2015;160(2):255–60.
Sharapova SO, Chang EY, Guryanova IE, Proleskovskaya IV, Fedorova AS, Rutskaya EA, et al. Next generation sequencing revealed DNA ligase IV deficiency in a “developmentally normal” patient with massive brain Epstein-Barr virus-positive diffuse large B-cell lymphoma. Clin Immunol. 2016;163:108–10.
Jiang J, Tang W, An Y, Tang M, Wu J, Qin T, et al. Molecular and immunological characterization of DNA ligase IV deficiency. Clin Immunol. 2016;163:75–83.
Fadda A, Butt F, Tomei S, Deola S, Lo B, Robay A, et al. Two hits in one: whole genome sequencing unveils LIG4 syndrome and urofacial syndrome in a case report of a child with complex phenotype. BMC Med Genet. 2016;17(1):84.
Felgentreff K, Baxi SN, Lee YN, Dobbs K, Henderson LA, Csomos K, et al. Ligase-4 deficiency causes distinctive immune abnormalities in asymptomatic individuals. J Clin Immunol. 2016;36(4):341–53.
Brunet BA, Dave N. Unique heterozygous presentation in an infant with DNA ligase IV syndrome. Ann Allergy Asthma Immunol. 2017;119(4):379–80.
Dard R, Herve B, Leblanc T, de Villartay JP, Collopy L, Vulliami T, et al. DNA ligase IV deficiency: immunoglobulin class deficiency depends on the genotype. Pediatr Allergy Immunol. 2017;28(3):298–303.
Cifaldi C, Angelino G, Chiriaco M, Di Cesare S, Claps A, Serafinelli J, et al. Late-onset combined immune deficiency due to LIGIV mutations in a 12-year-old patient. Pediatr Allergy Immunol. 2017;28(2):203–6.
Yue T, Li JG, Zhou ZX, Hou J, Xu YJ, Liu R, et al. LIG4 syndrome: a report of four cases and literature review. Chin J Pediatr. 2019;57(3):217–21.
Taskiran EZ, Sonmez HE, Kosukcu C, Tavukcuoglu E, Yazici G, Esendagli G, et al. A novel missense LIG4 mutation in a patient with a phenotype mimicking Behcet's disease. J Clin Immunol. 2019;39(1):99–105.
Monies D, Abouelhoda M, Assoum M, Moghrabi N, Rafiullah R, Almontashiri N, et al. Lessons learned from large-scale, first-tier clinical exome sequencing in a highly consanguineous population. Am J Hum Genet. 2019;104(6):1182–201.
Ding Y, Zhou L, Xia Y, Wang W, Wang Y, Li L, et al. Reference values for peripheral blood lymphocyte subsets of healthy children in China. J Allergy Clin Immunol. 2018;142(3):970–3.
Plowman PN, Bridges BA, Arlett CF, Hinney A, Kingston JE. An instance of clinical radiation morbidity and cellular radiosensitivity, not associated with ataxia-telangiectasia. Br J Radiol. 1990;63(752):624–8.
Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, et al. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000;5(6):993–1002.
Ashton JJ, Andreoletti G, Coelho T, Haggarty R, Batra A, Afzal NA, et al. Identification of variants in genes associated with single-gene inflammatory bowel disease by whole-exome sequencing. Inflamm Bowel Dis. 2016;22(10):2317–27.
Felgentreff K, Perez-Becker R, Speckmann C, Schwarz K, Kalwak K, Markelj G, et al. Clinical and immunological manifestations of patients with atypical severe combined immunodeficiency. Clin Immunol. 2011;141(1):73–82.
Uhlig HH, Schwerd T, Koletzko S, Shah N, Kammermeier J, Elkadri A, et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology. 2014;147(5):990–1007.
Girard PM, Kysela B, Harer CJ, Doherty AJ, Jeggo PA. Analysis of DNA ligase IV mutations found in LIG4 syndrome patients: the impact of two linked polymorphisms. Hum Mol Genet. 2004;13(20):2369–76.
Riballo E, Doherty AJ, Dai Y, Stiff T, Oettinger MA, Jeggo PA, et al. Cellular and biochemical impact of a mutation in DNA ligase IV conferring clinical radiosensitivity. J Biol Chem. 2001;276(33):31124–32.
O'Driscoll M, Jeggo PA. CsA can induce DNA double-strand breaks: implications for BMT regimens particularly for individuals with defective DNA repair. Bone Marrow Transplant. 2008;41(11):983–9.
Acknowledgments
The authors thank the patients and their families for their cooperation. We thank all the members of the research group.
Funding
This study was supported by the National Natural Science Foundation of China (81471482, 81373221) and Children’s Hospital of Fudan University Funding (EK1125180110, EK112520180202).
Author information
Authors and Affiliations
Contributions
Bijun Sun and Qiuyu Chen drafted the manuscript. Ying Wang and Danru Liu completed the experimental section. Jia Hou, Wenjie Wang, Wenjing Ying, Xiaoying Hui, Qinhua Zhou contributed to study design and data collection. Jinqiao Sun and Xiaochuan Wang gave academic feedback and revised the manuscript. All authors have reviewed the final manuscript and agreed to be accountable for the work.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of the Children’s Hospital of Fudan University. The patients and their parents provided written informed consent for enrollment in this study.
Consent for publication
Not applicable.
Competing interests
All the authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Immune index of patients with LIG4 mutations.
Additional file 2: Table S2.
Clinical and immunophenotype comparison between genotypes p.R814X/p.K424Rfs*20 and p.R278/p.K424Rfs*20.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sun, B., Chen, Q., Wang, Y. et al. LIG4 syndrome: clinical and molecular characterization in a Chinese cohort. Orphanet J Rare Dis 15, 131 (2020). https://doi.org/10.1186/s13023-020-01411-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-020-01411-x