
Abstract
Objective
To summarize and extend the phenotypic characterization of Multiple Congenital Anomalies-Hypotonia-Seizures Syndrome, and to discuss genotype-phenotype correlations.
Methods
Collecting clinical information of 17 patients with pathogenic variants in PIGN, PIGA, and PIGT. Genetic studies were performed on all patients.
Results
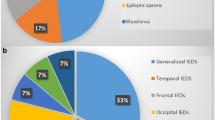
There were 7 patients with 15 PIGN mutations (one patient carrying 3 mutations), 8 patients with 8 PIGA mutations, and 2 patients with 5 PIGT mutations (one patient carrying 3 mutations). All patients had epilepsy and developmental delay, with 71% of them showed hypotonia. And among these patients’ various seizure types, the focal seizure was the most common one. Eighty-two percent patients showed a significant relationship between seizures and fever. Serum ALP was elevated in one patient with PIGN mutations and in two patients with PIGA mutations. Brain MRI showed enlarged subarachnoid space in 56% of patients. Some other different characteristics had also been found in our patients: First, atypical absence seizures presented in three patients with PIGN mutations; Second, diffuse slow waves mixed with focal or multifocal discharges of interictal EEG in 88% cases with PIGA-deficient; Third, phenotypes of seven out of eight patients with PIGA mutations were difficult to be classified as severe or less severe group; Last, mild neurological symptoms and developmental status rather than severe conditions occurred in one patient with PIGT mutations.
Conclusion
With epilepsy, developmental delay, and/or hypotonia as common features, the knowledge of MCAHS in terms of phenotype and genotype has been expanded. In cases with PIGN-deficient, we expanded the types of atypical absence seizures, and described one patient with elevated serum ALP. Focal seizures with diffuse slow waves mixed with focal or multifocal discharges on EEG rather than infantile spasms with hypsarrhythmia, which as previously reported were often seen in our patients with PIGA mutations. The classifications of phenotypes caused by PIGA mutations should be more continuous than discrete. The mild phenotype of one patient with PIGT mutations expanded the clinical presentation of MCAHS3.
Similar content being viewed by others


Introduction
Glycosylphosphatidylinositol (GPI) anchoring is a highly conserved process that enables proteins to attach to the cell surface membrane [1, 2]. Close to 30 genes have been identified, that are involved in the biosynthesis and remodeling of GPI-anchored proteins (GPI-APs), and variants in nearly 20 genes are associated with human diseases [3,4,5]. The function of GPI-related proteins is impaired due to defects in GPI anchored biosynthesis pathway, which have caused broad clinical phenotypes. The Multiple Congenital Anomalies-Hypotonia-Seizures Syndrome (MCAHS) is a clinically and etiologically heterogeneous disorder caused by mutations in the phosphatidylinositol glycan family, featuring hypotonia, seizures, multiple congenital anomalies, and delayed or lacking psychomotor development [5].
In 2012, the homozygous mutations of phosphatidylinositol glycan biosynthesis class N protein (PIGN, OMIM: 614080) was initially reported to lead to MCAHS1 in seven patients from a large consanguineous Israeli-Arab family [5]. Then, phosphatidylinositol glycan biosynthesis class A protein (PIGA, OMIM: 300868) and phosphatidylinositol glycan biosynthesis class T protein (PIGT, OMIM: 615398), in turn, were found to be responsible for MCAHS2 and MCAHS3 [6, 7]. In addition to PIGA being an X-linked recessive inheritance, both PIGN and PIGT were consistent with the characteristics of autosomal recessive inheritance.
PIGN controls the addition of phosphoethanolamine to the first mannose in GPI [8]. To date, a total of 29 patients with 22 different variants have been reported [5, 9,10,11,12,13,14,15,16,17,18,19]. Nineteen patients were diagnosed with MCAHS1 and 10 were diagnosed with Fryns syndrome which features were congenital diaphragmatic hernia (CDH), dysmorphic facial features, pulmonary hypoplasia, and other various possible internal malformations [5, 9,10,11,12,13,14,15,16,17,18,19]. CDH and associated pulmonary hypoplasia were responsible for early death in most patients with Fryns syndrome [9, 16, 18]. Because the patients with Fryns syndrome died during pregnancy or within a few days of birth, epilepsy and developmental delay were not observed in these patients [9, 16, 18]. MCAHS1 and Fryns syndrome share overlapping clinical features. The human PIGA maps to chromosome Xp22.2. Somatic PIGA mutations have been identified in many patients with paroxysmal nocturnal hemoglobinuria [20, 21]. Subsequently, germline PIGA mutations were associated with a spectrum of X-linked disorders containing MCAHS2 [6]. Up to now, 25 male patients carrying 15 different PIGA variants have been reported [6, 22,23,24,25,26,27,28,29,30,31,32,33,34]. PIGT encodes transaminase subunits involved in the GPI anchor binding to the protein at step 11 [35]. So far, 29 patients carrying 15 different PIGT variants have been reported [7, 36,37,38,39,40,41,42,43]. Since PIGN, PIGA, and PIGT are widely expressed in different tissues, clinical phenotypes of MCAHS are often complex and diverse.
Here, we present the clinical, biochemical and molecular characteristics of 17 new cases with MCAHS, in which the exome sequencing has revealed mutations in PIGN, PIGA, and PIGT.
Materials and methods
Ethics statement
This study was approved by the Biomedical Research Ethical Committee of Peking University First Hospital. The individuals’ parents in this manuscript have given the written informed consents to publish the case details.
Patients
Seventeen patients from 17 unrelated families were recruited. Several clinical data were analyzed. Electroencephalogram (EEG), brain magnetic resonance imaging (MRI), and biochemical studies were performed in all patients. Development assessment was also performed based on intelligence tests (Wechsler or Gesell intelligence scales) or clinical judgment and parents’ questionnaires.
Genetic analysis
Genetic analysis of PIGN, PIGA, and PIGT was performed as described by Maydan et al., Johnston et al., and Kvarnung et al. [5,6,7]. We also predicted the functional alteration of the mutations using polymorphism phenotyping-2 (PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/www/SIFT_seq_submit2.html) and Mutation Taster (http://www.mutationtaster.org/).
Results
Clinical features of patients with the PIGN/A/T mutations
The clinical information of individuals with PIGN, PIGA, and PIGT mutations is summarized in Table 1.
Phenotype of PIGN mutation
One patient (patient 4) out of seven patients had a history of preterm birth due to premature rupture of membranes. The other six patients were delivered at full term. The first seizures began between 3 months and 1 year. Focal seizures occurred in five patients (patient 1, 2, 4, 5, and 7), and other seizure types included atypical absence (3/7), myoclonic seizures (2/7), partial secondarily generalized tonic-clonic seizures (PGTCS) (1/7), and generalized tonic-clonic seizures (GTCS) (1/7). For five patients (patient 3, 4, 5, 6, and 7), the first seizures occurred in the course of febrile illness, after which febrile or afebrile seizures developed. Patient 2 had the first seizures during a hot bath, followed by focal seizures occurring about 3–4 times a month, almost always in the course of febrile illness. Although seizures of patient 1 developed without fever, almost all the subsequent seizures appeared during fever period. Facial deformities presented in six patients while other organ malformations were seen in three. Relevant facial and organ features of patient 1 were not available. Except that patient 1 who had no available results, five patients showed hypotonia.
Initial interictal EEG showed multifocal discharges in two patients (case 4 and 6). Patient 2 first showed discharges in left frontal and central regions, and had then been brought under control by the 5-month treatment of multiple antiepileptic drugs (AEDs). Initial interictal EEG of patient 1, 3, 5, and 7 were normal, however with only patient 5 exhibiting generalized discharges in re-examination 1 year later. The brain MRI of four patients showed enlarged subarachnoid space. In addition, other abnormalities, including enlarged ventricles (3/7), thin corpus callosum (1/7), slightly delayed myelin sheath (1/7), and cerebellar vermis dysplasia (1/7), were also present. By contrast, patient 2 and patient 6 showed neither dysmorphic features nor abnormalities in brain MRI. Serum alkaline phosphatase (ALP) was elevated in patient 4 (369 IU/L, normal range: 161.8–296.4 IU/L). The expression levels of total GPI-APs, CD16, CD24, and CD59 in peripheral blood granulocytes of patient 3 (the only patient tested) decreased to 34%, 28.9%, 19.1%, and 61.6% of normal controls, respectively.
In terms of drug treatments, seizures of six patients were refractory to AEDs, while those of patient 2 were controlled by levetiracetam. As for pyridoxine treatment for four patients, patient 2 was first treated orally with pyridoxine (90 mg/day) without effect at the age of one, and then at the aged of three seizures were controlled by AEDs. Also treated with pyridoxine (40 mg/day) combined with levetiracetam at the age of four, the condition of patient 3 failed to be controlled. Both patients 4 (90 mg/day) and 5 (40 mg/day) were treated with pyridoxine for nearly 4 months (at the last follow-up) without seizures at the age of four. With the two patients’ seizure frequency of once every 4–5 months, the efficacy of pyridoxine was difficult to assess.
Five patients showed severe developmental delay, particularly in language, while two patients were moderate. Patient 1 and 3 showed severe delayed intelligence and language with normal motor ability.
Phenotype of PIGA mutation
Among the eight male patients derived from 8 unrelated families (patient 8–15), patient 11 had a history of preterm labor and two patients showed perinatal abnormalities including anoxia or meconium-stained amniotic fluid. Patient 10 and 11 were diagnosed with congenital megacolon at the 28th days and the 13th days after birth, respectively. Convulsion occurred between 1 month and 28 days and 10 months. Focal seizures occurred in all patients. Other seizure types included PGTCS (1/8), GTCS (1/8), myoclonic seizure (1/8), absence seizure (1/8), and epileptic spasm (1/8), all of which could exist simultaneously in the same individual. In addition, three patients (patient 9, 10, and 11) had a history of status epilepticus. The first seizures in four patients occurred during a febrile episode, followed by febrile or afebrile seizures, mostly occurring during fever. The seizures of two patients showed a significant relationship with fever except for the first episode. The onset of seizures in patients 10 and 13 did not suggest a specific relationship with fever, but the seizures of patient 10 was aggravated during the course of febrile illness at times.
Physical examination data were not available in patient 15. In the remaining seven patients, three patients (patients 9, 10, and 11) showed facial dysmorphisms, with patient 9 also showing a foot deformity and an umbilical hernia. A cafe-au-lait-spot was observed on the back of patient 11. Except for two patients (patient 14 and 15) without available results, hypotonia could be seen in four patients. EEG findings were obtained from all eight patients. The initial interictal EEG showed hypsarrhythmia in patient 13. Other seven patients first demonstrated diffuse slow waves mixed with focal or multifocal discharges. Among the seven patients with brain MRI results, two patients were normal. While the most frequent abnormality was enlarged subarachnoid space (4/7), with others including enlarged ventricle (2/7), white-matter immaturity (1/7), and deeper brain sulcus (1/7). The above abnormalities could exist simultaneously in the same individual. Serum ALP level was mildly elevated (277 IU/L) in patient 10 at 6 months of age (normal range: 50–240 IU/L). As for patient 13, multiple results of serum ALP levels showed mild elevated (263 IU/L, normal range: 50–240 IU/L). The serum ALP levels of the other six patients were normal or without data.
Seizures of six patients were refractory to AEDs. In the other two patients, the seizures of patient 9 were controlled by the combination treatment of levetiracetam, valproic acid, and topiramate at the age of 16 months. Similarly, as for patient 14, the seizures were eventually controlled by valproate and levetiracetam at about 2 years of age. Three patients (patient 10, 11, and 13) were treated with pyridoxine. Among them, patient 10 and 11 were treated with pyridoxine intravenously for 6 days at the age of 6 months (100 mg/day) without effects, while patient 13 was treated with pyridoxine at the age of about 5 months, whose seizures remained uncontrolled no matter by the administration of oral (40 mg/day) or intravenous (100 mg/day). Besides, ketogenic diet was used in three patients during the course without obvious effects. All patients showed severe developmental delay, with patient 13 showing severe delayed intelligence and language with normal motor ability.
Phenotype of PIGT mutation
Patient 16
The male patient was the child of non-consanguineous Chinese parents. The first seizures occurred during fever at the age of 3, lasting for approximately 1 minute, with 7–8 times per year. Then the improved afebrile seizures occurred at the age of 9, uncontrolled by the combination of levetiracetam and oxcarbazepine. From the age of 9, he gradually became unsteady in walking and prone to falls with a worsen tendency. He is currently unable to stand up from sitting independently. Physical examination showed bilateral knee hyperreflexia, normal muscle strength and muscle tone, without facial or organ dysmorphism. EEG showed discharges in the left anterior and middle temporal areas during sleep at 9 years of age. After AEDs treatment, multiple EEG results were normal. Brain MRI and metabolic analyses in urine and plasma showed normal. Since the seizure onset, his motor development has been severely delayed, but intelligence and language are normal.
Patient 17
The male patient was the first child of non-consanguineous healthy Chinese parents. Pregnancy, labor and early infancy were normal. Motor milestones, like raising head and rolling over, were achieved later than infants of the same age. Seizures started at the age of 4 months, presenting as left lower limb shaking once or many times during the wake-up of 10–12 times per day. There was no significant correlation between seizures and fever. At about 5 months, the above seizures still existed without therapy, with the frequency increasing to 30–40 times per day. The seizures were reduced but not controlled by the therapy of valproate, levetiracetam, lamotrigine, and oxcarbazepine. Besides seizures, he showed startle responses to unexpected sound stimuli from childhood. Physical examination showed a cafe-au-lait-spot on his right knee. He had severe axial hypotonia, nasal bridge collapse, arched palatal, and dysplastic ears. At 10 months old, EEG showed multiple discharges and brain MRI showed enlarged subarachnoid space. With severe developmental delay, he was unable to stand or sit, having the unconscious pronunciation at 1 year old (the last follow-up).
Molecular results
The details of molecular variants identified in PIGN, PIGA, and PIGT were summarized in Table 2, including the mutations of the genes with pedigrees, the frequency of variants among the normal population, and the scores in the prediction tool. And among the other variants, with only the PIGA mutation c. 1370C > T (p. A457V) predicted as “neutral” by SIFT, all the remaining were predicted to be “probably damaging”, “deleterious”, and “disease causing” to the protein structure identified by Polyphen-2, SIFT and Mutation Taster, respectively.
PIGN mutations
Fifteen variants from seven patients were identified with PIGN mutations (NM_012327), including 8 missense mutations, 4 nonsense mutations (one variant has been reported) [16], one synonymous mutation (has been reported) [15], one splicing site mutation, and one deletion mutation. Among all patients, case 4 had 3 mutations. Two mutations [c.694A > T (p. K232X) and c.963G > A (p. Q321Q)] had been reported previously [15, 16]. All variants were validated by Sanger sequencing confirming their parental inheritance.
PIGA mutations
Mutation analysis identified seven different hemizygous PIGA mutations (NM_002641) among eight patients, all of which were missense mutations. Of them, R119Q presented in two patients here and had been reported before [34]. Seven of the mutations were inherited from their mothers, and only one was de novo.
PIGT mutations
Five PIGT variants were identified in two sporadic patients (NM_015937), while case 16 carried 3 mutations. Among these, three mutations were missense mutations, while the other two were deletion mutation and deletion insertion mutation. None of the mutations had been previously reported. The c.469 T > G variant was a missense transition from phenylalanine to valine. Additionally, it was predicted to be deleterious by three algorithms (PolyPhen-2, SIFT, and Mutation Taster). The c.1120A > G variant was also predicted to be highly damaged to the protein structure. The c.514C > T variant is a missense transition from arginine to cysteine at residue 172 of the protein, predicted as deleterious by the above predictive software packages.
Discussion
In 17 patients with a congenital disorder of MCAHS, the whole exome sequencing revealed pathogenic variants of the PIGN, PIGA, and PIGT. Among them, one patient had 3 PIGN mutations, and another patient had 3 PIGT mutations. There was no previous report of patients with 3 PIGN, PIGA, or PIGT mutations simultaneously. In the wide variation of the MCAHS phenotypic spectrum, our patients differed significantly in addition to the several similarities with the previously reported cases, broadening the scope of knowledge on inherited GPI deficiency.
Early onset seizures appeared to be a common feature of the 83 patients previously reported with PIGN, PIGA, and PIGT mutations. Based on previously reported cases, the most common type in patients with PIGN mutations [5, 9,10,11,12,13,14,15,16,17,18,19] was focal seizure, which also presented in our patients. Three patients with PIGN mutations showed atypical absence, which had not been seen in previous reports. By summarizing the PIGA mutations cases reported, myoclonic seizure was the most common type (10/19 patients with available data) [6, 22,23,24,25,26,27,28,29,30,31,32,33,34]. However, Tarailo-Graovac et al. [24] who described one case and summarized 10 cases reported previously found that infantile spasms with hypsarrhythmia on EEG was the hallmarks of these patients. Different from the above reports, our eight cases with PIGA mutations were characterized by neither epileptic spasms nor myoclonic seizures, but by focal seizures (100%, 8/8) and status epilepticus (38%, 3/8). Besides, EEGs, were more of diffuse slow waves mixed with focal or multifocal discharges (7/8), but did not reach the degree of hypsarrhythmia. In the previous reports, myoclonic seizure (17/25 patients with available data) was the most common type in PIGT mutations patients, without epileptic spasms presented [7, 36,37,38,39,40,41,42,43]. In our cohort, focal seizures, epileptic spasms, and myoclonic seizures occurred in both patients with PIGT mutations.
Intractable seizures occurred in the majority reported previously (60/72 patients with available data) and here [5,6,7, 9,10,11,12,13,14,15,16,17,18,19, 22,23,24,25,26,27,28,29,30,31,32,33,34, 36,37,38,39,40,41,42,43]. Chiyonobu et al. [44] hypothesized that the seizures in these patients were due to a defect in intraneuronal pyridoxal phosphate (PLP), a cofactor of γ-amino butylic acid (GABA) synthase, which was caused by a loss of membrane-anchored ALP, leading to a shortage of GABA. Pyridoxine treatment was reported to be effective for seizures in some patients with inherited GPI deficiencies, such as PIGV or PIGO mutations [45,46,47]. However, two patients with PIGN mutations and three patients with PIGA mutations in this study failed to respond to pyridoxine treatment. Up to now, only two patients with PIGA mutations and one patient with PIGT mutations were reported to receive pyridoxine treatment [30, 34, 40]. In addition, two patients with PIGT mutations have also been treated with PLP [36, 40]. Neither pyridoxine nor PLP were effective, although the number of cases was limited. It was unclear whether genetic heterogeneity played a role in only some individuals’ responsiveness to pyridoxine but not in others. Therefore, more patients with intractable seizures should be treated with pyridoxine or PLP to determine whether such treatment might be effective for some patients with PIGN, PIGA, and PIGT mutations.
In terms of development, among previously reported cases, except for two patients with moderate delay carrying PIGN and PIGA mutations [17, 30], the others had severe delay or early death [5,6,7, 9,10,11,12,13,14,15,16,17,18,19, 22,23,24,25,26,27,28,29,30,31,32,33,34, 36,37,38,39,40,41,42,43]. In our cohort, there are two patients with moderate delay carrying PIGN mutations. In addition, patient 16 with PIGT mutations showed mild developmental delay without dysmorphisms or severe intellectual disability. Moreover, the onset of seizures was relatively late, and the findings of brain MRI were normal. To date, he was the only PIGT mutations patient having neither facial nor other organ defects with the latest seizure onset age. Overall, the clinical phenotype of patient 16 expanded the clinical presentation of MCAHS3.
Hyperphosphatemia is a characteristic symptom of some GPI deficiencies, including PIGO, PIGW, PGAP2, etc. [48,49,50]. Such elevations have not been reported in the previous cases of PIGN mutations. However, patient 4 with 3 PIGN mutations, showed high serum ALP levels. There were no previous reports of patients with 3 PIGN mutations simultaneously which might have a great influence on the function of GPI. Therefore, the increased level of serum ALP in patient 4 might be caused by the dysfunction of GPI protein due to the insufficient residual function of PIGN, based on the mechanism of PIGA mutations. Mildly elevated serum ALP was also reported in more than half of the patients with PIGA mutations (9/15 patients with available data), including two patients here [6, 22,23,24,25,26,27,28,29,30,31,32,33,34]. A possible biological explanation was that when cell surface GPI-APs, such as ALP, could not be modified by GPI, they were secreted as soluble non-GPI-anchored proteins [50]. The elevated serum ALP might serve as a diagnostic clue rather than a criterion. In contrast, hypophosphatasia was a particularly distinctive feature in patients with PIGT mutations (7/13 patients with available data) [7, 36,37,38,39,40,41,42,43]. Murakami et al. [50] considered that abnormal GPI transaminase resulted in the difficulty in hydrolysis of ALP precursor protein, leading to the degradation of most of the precursor protein in cells and the decrease of serum ALP. However, the feature of hypophosphatasia was not found in our two PIGT mutations cases. In addition, the level of GPI anchored proteins on the granulocyte surface decreased in patients with GPI deficiencies, such as CDl6, CD24, CD59, etc. [5, 15]. In this study, flow cytometry was performed on only one case with PIGN mutations. The result showed that the expression levels of total GPI-APs CD16, CD24, and CD59 in peripheral blood granulocytes were decreased, which confirmed the effect of mutation on PIGN function.
By summarizing the previously reported cases, in patients with PIGN mutations, white matter immaturity and thinning of the corpus callosum were the most common brain MRI abnormities (38%, 8/21 patients with available data) [5, 9,10,11,12,13,14,15,16,17,18,19]. Brain MRI showed cerebellar atrophy in 76% of the PIGT-deficient patients (10/14 patients with available data), which also accounted for the majority of patients with PIGA mutations (25%, 4/16 patients with available data) [6, 7, 22,23,24,25,26,27,28,29,30,31,32,33,34, 36,37,38,39,40,41,42,43]. Different from the previous reports, enlarged subarachnoid space was the most common brain MRI abnormality in the patients we reported, regardless of which type of mutations.
Up to now, different PIGN, PIGA, and PIGT disease-causing variants have been reported in 22, 15, and 15 mutation sites, respectively [5,6,7, 9,10,11,12,13,14,15,16,17,18,19, 22,23,24,25,26,27,28,29,30,31,32,33,34, 36,37,38,39,40,41,42,43]. Here, we identified 15 PIGN variants (one patient carrying 3 mutations), 8 PIGA variants, and 5 PIGT variants (one patient carrying 3 mutations). In the case of patient 4 with PIGN mutations, there were two paternal mutations and one maternal. The pathogenicity of the maternal synonymous mutation site c.963G > A has been reported and proved to have actual effects of on splicing [15]. The presence of 3 mutations all affected the function of GPI-APs and made the phenotype of patient 4 more severe, which was characterized by a history of preterm labor, intractable seizures, severe developmental delay, multiple facial and other organ deformities, and especially elevated serum ALP level. Previous reports had suggested a genotype-phenotype correlation of PIGN mutations [14, 18]. The clinical severity of the cases reported to date seemed to correlate with the predicted functional severity of the mutations in PIGN [14]. Specifically, the fatal biallelic truncating variant of PIGN was identified in patients with a severe clinical course, including early onset of intractable seizures and death in utero or shortly after birth. Our patients did not carry biallelic loss of function variants and showed the milder phenotype without major visceral congenital anomalies. It was further proved that the biallelic mutations predicting truncated proteins with presumed loss of function variants were likely responsible for the severe phenotype of PIGN mutations.
As for the variants of PIGA, to date, a total of seven patients carrying c.1234C > T have been reported, with a very similar phenotype [6, 21, 29]. This suggested that the phenotype might correlate with genotype and the residual functional activity of the PIGA protein. However, though our two patients with the same c.356G > A mutation had similar onset of seizures, AEDs resistance, and severe developmental delay, multiple organ anomalies and hypotonia only occurred in one of them. This variability indicated that the phenotype was influenced by other factors in addition to genotypes. In general, the phenotypic spectrum caused by PIGA germline mutations were classified into two types previously, severe and less severe [30]. According to phenotype and genotype, 16 out of 25 patients reported previously were divided into the severe group, as was our patient 10 [6, 22,23,24,25,26,27,28,29,30,31,32,33,34]. However, in the rest of the patients, only a few (Patient IV-2 by Belet et al., 2013; patient 3 and patient 4 by Kato et al., 2014; patient 1 and patient 2 by Joshi et al., 2016) characteristic of the less severe form [26, 27, 30]. The remaining 11 patients reported here, and previously, their phenotypes could hardly be divided into severe or less severe group. For example, except for patient 10, though the other seven patients reported here presented with intractable seizures (5/7), severe developmental delay (7/7), multiple organ malformations (2/6), hypotonia (3/5), lack of dysmorphism, elevated serum ALP levels precluded them to be included into severe form. And obviously, they did not belong to the less severe group. It indicated that the phenotypic range of PIGA mutations was more continuous than discrete.
Additionally, previous reports had shown that PIGT was expressed at the highest level in the central nervous system, which might explicate that the neurological defect was the core features and the most severe [38, 39]. Abnormalities in other organ systems might not be considered as a necessary diagnostic factor for PIGT mutations [38, 39]. However, mild neurological symptoms and developmental status were detected in our proband (patient 16), with motor ability decreasing after seizures onset. In addition to the two missense mutations, patient 16 also carried a paternal deletion insertion mutation, both of which involved three bases. Thus, each of the three mutations altered a single amino acid in GPI-APs and was predicted by three algorithms (PolyPhen-2, SIFT, and Mutation Taster) to be pathogenic. One possible explanation for its lighter phenotype was that amino acids altered by PIGT mutations had less effect on GPI-APs function. Moreover, whether these three mutations or one of them would always be related to the milder phenotype needs more cases to be verified. In short, PIGT mutations could also cause mild neurological manifestations without other malformations.
Conclusions
We expand our knowledge of MCAHS in terms of phenotype and genotype. There were 7 patients with 15 PIGN mutations, 8 patients with 8 PIGA mutations, and 2 patients with 5 PIGT mutations. All patients had epilepsy and developmental delay, and 71% of them showed hypotonia. Focal seizure was the most common type in all cases. Eighty-two percent of patients showed a significant relationship between seizures and fever. Brain MRI abnormalities showed enlarged subarachnoid space in 56% patients. Among patients with PIGN mutations, atypical absence seizures had not been documented in previous reports. We have described here the only patient carrying PIGN mutations with elevated serum ALP. Our cases with PIGA mutations were characterized by focal seizures with diffuse slow waves mixed with focal or multifocal discharges on EEG rather than infantile spasms with hypsarrhythmia [24]. Except for one with PIGA mutations classified as severe, it was difficult to classify the phenotypes of our remaining seven patients as severe or less severe group, which indicated that phenotypes were more continuous than discrete. Mild neurological symptoms and developmental status of one patient with PIGT mutations expanded the clinical presentation of MCAHS3.
Availability of data and materials
The datasets generated and/or analyzed during the current study are not publicly available due to patient data safety restrictions but are available from the corresponding author on reasonable request.
Abbreviations
- GPI:
-
Glycosylphosphatidylinositol
- GPI-APs:
-
GPI-anchored proteins
- MCAHS:
-
Multiple congenital anomalies-hypotonia-seizures syndrome
- PIGN:
-
Phosphatidylinositol glycan biosynthesis class N protein
- PIGA:
-
Phosphatidylinositol glycan biosynthesis class A protein
- PIGT:
-
Phosphatidylinositol glycan biosynthesis class T protein
- PGTCS:
-
Partial secondarily generalized tonic-clonic seizure
- GTCS:
-
Generalized tonic-clonic seizure
- EEG:
-
Electroencephalogram
- MRI:
-
Magnetic resonance imaging
- AEDs:
-
Antiepileptic drugs
- ALP:
-
Alkaline phosphatase
- PLP:
-
Pyridoxal phosphate
- GABA:
-
γ-amino butylic acid
- CDH:
-
Congenital diaphragmatic hernia
References
Muñiz M, Riezman H. Trafficking of glycosylphosphatidylinositol anchored proteins from the endoplasmic reticulum to the cell surface. J Lipid Res. 2016;57(3):352–60.
Ng BG, Freeze HH. Human genetic disorders involving glycosylphosphatidylinositol (GPI) anchors and glycosphingolipids (GSL). J Inherit Metab Dis. 2015;38(1):171–78.
Wu TH, Peng J. Progress of PIG/PGAP gene and related phenotype. Chin Appl Clin J Pediatr. 2019;34(9):714–17.
Nguyen TTM, Murakami Y, Sheridan E, Ehresmann S, Rousseau J, St-Denis A, et al. Mutations in GPAA1, encoding a GPI transamidase complex protein, cause developmental delay, epilepsy, cerebellar atrophy, and osteopenia. Am J Hum Genet. 2017;101(5):856–65.
Maydan G, Noyman I, Har-Zahav A, Neriah ZB, Pasmanik-Chor M, Yeheskel A, et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet. 2011;48(6):383–9.
Johnston JJ, Gropman AL, Sapp JC, Teer JK, Martin JM, Liu CF, et al. The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am J Hum Genet. 2012;90(2):295–300.
Kvarnung M, Nilsson D, Lindstrand A, Korenke GC, Chiang SC, Blennow E, et al. A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J Med Genet. 2013;50(8):521–8.
Freeze HH, Eklund EA, Ng BG, Patterson MC. Neurology of inherited glycosylation disorders. Lancet Neurol. 2012;11(5):453–66.
Brady PD, Moerman P, De Catte L, Deprest J, Devriendt K, Vermeesch JR. Exome sequencing identifies a recessive PIGN splice site mutation as a cause of syndromic congenital diaphragmatic hernia. Eur J Med Genet. 2014;57(9):487–93.
Couser NL, Masood MM, Strande NT, Foreman AK, Crooks K, Weck KE, et al. The phenotype of multiple congenital anomalies-hypotonia-seizures syndrome 1: Report and review. Am J Med Genet A. 2015;167A(9):2176–81.
Jezela-Stanek A, Ciara E, Piekutowska-Abramczuk D, Trubicka J, Jurkiewicz E, Rokicki D, et al. Congenital disorder of glycosylphosphatidylinositol (GPI)-anchor biosynthesisdThe phenotype of two patients with novel mutations in the PIGN and PGAP2 genes. Eur J Paediatr Neurol. 2016;20(3):462–73.
Khayat M, Tilghman JM, Chervinsky I, Zalman L, Chakravarti A, Shalev SA. A PIGN mutation responsible for multiple congenital anomalies–hypotonia–seizures syndrome 1 (MCAHS1) in an Israeli–Arab family. Am J Med Genet A. 2016;170A(1):176–82.
Pagnamenta AT, Murakami Y, Taylor JM, Anzilotti C, Howard MF, Miller V, et al. Analysis of exome data for 4293 trios suggests GPI-anchor biogenesis defects are a rare cause of developmental disorders. Eur J Hum Genet. 2017;25(6):669–79.
Thiffault I, Zuccarelli B, Welsh H, Yuan X, Farrow E, Zellmer L, et al. Hypotonia and intellectual disability without dysmorphic features in a patient with PIGN-related disease. BMC Med Genet. 2017;18(1):124.
Ohba C, Okamoto N, Murakami Y, Suzuki Y, Tsurusaki Y, Nakashima M, et al. PIGN mutations cause congenital anomalies, developmental delay, hypotonia, epilepsy, and progressive cerebellar atrophy. Neurogenetics. 2014;15(2):85–92.
McInerney-Leo AM, Harris JE, Gattas M, Peach EE, Sinnott S, Dudding-Byth T, et al. Fryns syndrome associated with recessive mutations in PIGN in two separate families. Hum Mutat. 2016;37(7):695–702.
Fleming L, Lemmon M, Beck N, Johnson M, Mu W, Murdock D, et al. Genotype-phenotype correlation of congenital anomalies in multiple congenital anomalies hypotonia seizures syndrome (MCAHS1)/PIGN-related epilepsy. Am J Med Genet A. 2016;170A(1):77–86.
Alessandri JL, Gordon CT, Jacquemont ML, Gruchy N, Ajeawung NF, Benoist G, et al. Recessive loss of function PIGN alleles, including an intragenic deletion with founder effect in La Réunion Island, in patients with Fryns syndrome. Eur J Hum Genet. 2018;26(3):340–9.
Nakagawa T, Taniguchi-Ikeda M, Murakami YA, Nakamura S, Motooka D, Emoto T, et al. A novel PIGN mutation and prenatal diagnosis of inherited Glycosylphosphatidylinositol deficiency. Am J Med Genet A. 2016;170A(1):183–8.
Keller P, Tremml G, Rosti V, Bessler M. X inactivation and somatic cell selection rescue female mice carrying a Piga-null mutation. Proc Natl Acad Sci USA. 1999;96(13):7479–83.
Keller P, Payne JL, Tremml G, Greer PA, Gaboli M, Pandolfi PP, et al. FES-Cre targets phosphatidylinositol glycan class a (PIGA) inactivation to hematopoietic stem cells in the bone marrow. J Exp Med. 2001;194(5):581–9.
Kim YO, Yang JH, Park C, Kim SK, Kim MK, Shin MG, et al. A novel PIGA mutation in a family with X-linked, early-onset epileptic encephalopathy. Brain Dev. 2016;38(8):750–4.
Swoboda KJ, Margraf RL, Carey JC, Zhou H, Newcomb TM, Coonrod E, et al. A Novel Germline PIGA Mutation in Ferro-Cerebro-Cutaneous Syndrome: A Neurodegenerative X-linked Epileptic Encephalopathy With Systemic Iron-Overload. Am J Med Genet A. 2014;164A(1):17–28.
Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S, Van Allen M, Rozmus J, Shyr C, et al. The genotypic and phenotypic spectrum of PIGA deficiency. Orphanet J Rare Dis. 2015;10:23.
van der Crabben SN, Harakalova M, Brilstra EH, van Berkestijn FM, Hofstede FC, van Vught AJ, et al. Expanding the Spectrum of phenotypes associated with Germline PIGA mutations: a child with developmental delay, accelerated linear growth, facial Dysmorphisms, elevated alkaline phosphatase, and progressive CNS abnormalities. Am J Med Genet A. 2014;164A(1):29–35.
Joshi C, Kolbe DL, Mansilla MA, Mason S, Smith RJ, Campbell CA. Ketogenic diet-a novel treatment for early epileptic encephalopathy due to PIGA deficiency. Brain Dev. 2016;38(9):848–51.
Belet S, Fieremans N, Yuan X, Van Esch H, Verbeeck J, Ye Z, et al. Early Frameshift mutation in PIGA identified in a large XLID family without neonatal lethality. Hum Mutat. 2014;35(3):350–55.
Trump N, McTague A, Brittain H, Papandreou A, Meyer E, Ngoh A, et al. Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis. J Med Genet. 2016;53(5):310–17.
Fauth C, Steindl K, Toutain A, Farrell S, Witsch-Baumgartner M, Karall D, et al. A recurrent germline mutation in the PIGA gene causes Simpson-Golabi-Behmel syndrome type 2. Am J Med Genet A. 2016;170A(2):392–402.
Kato M, Saitsu H, Murakami Y, Kikuchi K, Watanabe S, Iai M, et al. PIGA mutations cause early-onset epileptic encephalopathies and distinctive features. Neurology. 2014;82(18):1587–96.
Low KJ, James M, Sharples PM, Eaton M, Jenkinson S, Study DDD, et al. A novel PIGA variant associated with severe X-linked epilepsy and profound developmental delay. Seizure. 2018;56:1–3.
Xie LL, Song XJ, Li TY, Jiang L. A novel germline PIGA mutation causes early-onset epileptic encephalopathies in Chinese monozygotic twins. Brain Dev. 2018;40(7):596–600.
Yang JL, Wang Q, Zhuo QC, Tian HL, Li W, Luo F, et al. A likely pathogenic variant putatively affecting splicing of PIGA identified in a multiple congenital anomalies hypotonia-seizures syndrome 2 (MCAHS2) family pedigree via whole-exome sequencing. Mol Genet Genomic Med. 2018;6(5):739–48.
Lin WD, Chou IC, Tsai FJ, Hong SY. A novel PIGA mutation in a Taiwanese family with early-onset epileptic encephalopathy. Seizure. 2018;58:52–4.
Kinoshita T, Fujita M, Maeda Y. Biosynthesis, remodelling and functions of mammalian GPI-anchored proteins: recent progress. J Biochem. 2008;144(3):287–94.
Nakashima M, Kashii H, Murakami Y, Kato M, Tsurusaki Y, Miyake N, et al. Novel compound heterozygous PIGT mutations caused multiple congenital anomalies-hypotonia-seizures syndrome 3. Neurogenetics. 2014;15(3):193–200.
Lam C, Golas GA, Davids M, Huizing M, Kane MS, Krasnewich DM, et al. Expanding the clinical and molecular characteristics of PIGT-CDG, a disorder of Glycosylphosphatidylinositol anchors. Mol Genet Metab. 2015;115(2-3):128–40.
Skauli N, Wallace S, Chiang SC, Barøy T, Holmgren A, Stray-Pedersen A, et al. Novel PIGT Variant in Two Brothers: Expansion of the Multiple Congenital Anomalies-Hypotonia Seizures Syndrome 3 Phenotype. Genes. 2016;7(12):E108.
Yang L, Peng J, Yin XM, Pang N, Chen C, Wu TH, et al. Homozygous PIGT mutation Lead to multiple congenital anomalies-Hypotonia seizures syndrome 3. Front Genet. 2018;9:153.
Kohashi K, Ishiyama A, Yuasa S, Tanaka T, Miya K, Adachi Y, et al. Epileptic apnea in a patient with inherited glycosylphosphatidylinositol anchor deficiency and PIGT mutations. Brain Dev. 2018;40(1):53–7.
Mason S, Castilla-Vallmanya L, James C, Ian Andrews P, Balcells S, Grinberg D, et al. Case report of a child bearing a novel deleterious splicing variant in PIGT. Medicine. 2019;98(8):e14524.
Larsen KB, Bayat A, Møller RS, Maroun LL, Lund EL. First report of the neuropathological findings in a patient with leukodystrophy and compound heterozygous variants in the PIGT gene. Neuropathol Appl Neurobiol. 2019;45(7):732–5.
Bayat A, Knaus A, Juul AW, Dukic D, Gardella E, Charzewska A, et al. PIGT-CDG, a disorder of the glycosylphosphatidylinositol anchor: description of 13 novel patients and expansion of the clinical characteristics. Genet Med. 2019;21(10):2216–23.
Chiyonobu T, Inoue N, Morimoto M, Kinoshita T, Murakami Y. Glycosylphosphatidylinositol (GPI) anchor deficiency caused by mutations in PIGW is associated with west syndrome and hyperphosphatasia with mental retardation syndrome. J Med Genet. 2014;51(3):203–7.
Xue J, Li H, Zhang YH, Yang ZX. Clinical and genetic analysis of two Chinese infants with Mabry syndrome. Brain Dev. 2016;38(9):807–18.
Kuki I, Takahashi Y, Okazaki S, Kawawaki H, Ehara E, Inoue N, et al. Vitamin B6-responsive epilepsy due to inherited GPI deficiency. Neurology. 2013;81(16):1467–69.
Thompson MD, Killoran A, Percy ME, Nezarati M, Cole DE, Hwang PA. Hyperphosphatasia with neurologic deficit: a pyridoxine-responsive seizure disorder? Pediatr Neurol. 2006;34(4):303–7.
Krawitz PM, Murakami Y, Hecht J, Kruger U, Holder SE, Mortier GR, et al. Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am J Hum Genet. 2012;91(1):146–51.
Howard MF, Murakami Y, Pagnamenta AT, Daumer-Haas C, Fischer B, Hecht J, et al. Mutations in PGAP3 impair GPI-anchor maturation, causing a subtype of hyperphosphatasia with mental retardation. Am J Hum Genet. 2014;94(2):278–87.
Murakami Y, Kanzawa N, Saito K, Krawitz PM, Mundlos S, Robinson PN, et al. Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome. J Biol Chem. 2012;287(9):6318–25.
Acknowledgments
We thank the patients and their families for participating.
Funding
This work was supported by National Nature Science Foundation of China (81771393), Beijing Municipal Science & Technology Commission (Z171100001017125), and Beijing Natural Science Foundation (7202210).
Author information
Authors and Affiliations
Contributions
Ms. Xianru Jiao carried out the acquisition of data, analysis, interpretation of the data, and drafting and revising the manuscript; Ms. Jiao Xue and Pan Gong carried out the design of study, data collection, analysis and revising the manuscript. Dr. Xinhua Bao, Ye Wu, Yuehua Zhang and Yuwu Jiang carried out the acquisition of data and interpretation of data; Dr. Zhixian Yang conceptualized and designed the study in addition to the acquisition of data, analysis, interpretation of the data and revising the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Biomedical Research Ethical Committee of Peking University First Hospital. The individuals’ parents in this manuscript have given written informed consent to publish the case details.
Consent for publication
Not applicable.
Competing interests
The authors have stated that they had no interests which might be perceived as posing a conflict or bias.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jiao, X., Xue, J., Gong, P. et al. Analyzing clinical and genetic characteristics of a cohort with multiple congenital anomalies-hypotonia-seizures syndrome (MCAHS). Orphanet J Rare Dis 15, 78 (2020). https://doi.org/10.1186/s13023-020-01365-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-020-01365-0