
Abstract
Background
Infantile Sandhoff disease (ISD) is a GM2 gangliosidosis that is classified as a lysosomal storage disorder. The most common symptoms of affected individuals at presentation are neurologic involvement. Here we report clinical course and demographic features in a case series of infantile Sandhoff disease. Enzymatically and some genetically proven cases of ISD were extracted from the Iranian Neurometabolic Registry (INMR) in Children’s Medical Center, Iran, Tehran from December 2010 to December 2016.
Result
Twenty five cases of infantile SD (13 female, 12 male) were included in this study. The age range of patients was 9–24 months with a mean of 15.8 months. The consanguinity rate of parents affected families was about 80%. The mean age of patients at disease onset was 6.4 months and the mean age at diagnosis was 14 months. Patients were diagnosed with a mean delay of 7.8 months. Eleven of patients died due to aspiration pneumonia and intractable seizure. The most common features at presentation (92%) were developmental delay or regression in speech and cognitive domains. Cherry red spots were detected in 17 patients (68%). Organomegaly was detected only in two patients. Enzyme studies showed marked reductions of both Hexosaminidase A and B in all patients. HEXB gene mutation studies performed in eight patients identified 6 different mutations, which five of them were novel.
Conclusion
Infantile SD should be considered for each child presented with neurologic symptoms such as developmental delay and regression and cherry red spots in ophthalmic examination. Organomegaly is not a frequent clinical finding in infantile SD. Additionally; there are a genetic heterogenisity among Iranian patients.
Similar content being viewed by others

Background
Lysosomal storage disorders (LSDs) are a specific group of inborn errors of metabolism including more than fifthy different diseases caused by a structural defect or deficiency of lysosomal enzymes [1]. GM2 gangliosidosis is one group of LSDs that is classified into three types: Sandhoff (0 variant), Tay-Sachs (B variant) and GM2 activator deficiency (GM2A-AB variant). Mutations in HEXA, HEXB and GM2A genes that are inherited by autosomal recessive pattern lead to defective β-Hexosaminidase activity and accumulation of GM2 ganglioside in the intracellular organelles of visceral and neural cells [2]. Sandhoff disease (SD, OMIM 26880) due to a deficiency of β-Hexosaminidase activity (A and B units) was first described by Warren Tay in 1881 [3, 4]. The clinical manifestations of Tay-Sachs (OMIM 606869) and Sandhoff disease are the same and have been classified clinically into three forms of infantile, juvenile and adult. The clinical severity and age at disease onset are related to residual enzyme activity. Infantile SD presents with truncal hypotonia, muscle weakness, hyperacusis, developmental delay and regression, seizure and cherry red spots in ophthalmologic exam around 6 months of age. Hepatosplenomegaly, coarse facies and bone abnormality are less seen than Tay-Sachs disease. Death occurs by 3 years of age due to intractable seizure and aspiration pneumonia. The juvenile form of SD present between 2 and 10 years of age with dysarthria, ataxia, mental deterioration and seizures. Organomegaly and cherry red spots are uncommon. Adult form of SD is characterized by movement disorder, pyramidal and extra pyramidal signs and symptoms of lower motor neuron disease and supraneuclear ophtalmoplegia [2]. Molecular analysis and enzyme activity assessment are necessary to confirm the diagnosis of SD. There is no specific treatment for GM2 gangliosidosis but chaperone therapy with ketogenic diet and miglustat has been able to increase the cardiac function and reduce the frequency of seizures [3]. Due to the broad spectrum of clinical signs and symptoms of the disease and variety of mutations in the HEXB among the different races, herein we report the spectrum of clinical manifestations, clinical course and outcome of 25 cases of infantile SD presenting five novel mutations.
Methods
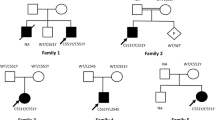
We stablished a bioregistry system in our hospital Children’s Medical Center hospital, Tehran, Iran in 2010. During 7 years clinical and laboratory data of more than 270 patients with different types of neurometabolic disorders has been registered. Twenty five enzymatically and genetically proven cases of SD were extracted from the Iranian Neurometabolic Registry (INMR) from December 2010 to December 2017. Early diagnosis was carried out according to clinical manifestations, physical and neurologic examinations, neuro-imaging findings followed by assessment of hexosaminidase enzyme activity in peripheral blood leukocyte. The diagnosis of SD was confirmed by molecular study in some patients. All parents entered the study with informed consent (Fig. 1).

Step by step evaluation of a child suspected to infantile Sandhoff disease
β-Hexosaminidase activity assessment
The activity of β-Hexosaminidase was assessed through dried blood spots (DBSs) on filter paper for all included patients. The blood spots on filter paper placed in 3 mm diameter. After incubation of the samples at 37 °C, the amount of enzyme activity achieved by compared the value of hydrolyzed protein with a standard sample [5].
Molecular study
Mutation analysis of HEXB gene was carried out on all coding exons and adjacent intron boundaries of the gene by PCR (Polymerase chain reaction) amplification and Sanger sequencing using standard methods.
Result
Demographic findings and clinical course
Twenty five cases of infantile SD (13 female, 12 male) enrolled in the study. The age of patients was in the range of 9–24 months with mean age of 15.8 months. The consanguinity rate of parents was about 80%. Familial history of previous SD was revealed in three patients and nine mothers announced unexplained death in other sibling. In addition, two mothers had previous unexplained abortion. The mean age of patients at the onset of clinical manifestations was 6.4 months and the mean at diagnosis was 14 months. On average, patients were diagnosed with a mean delay of 7.8 months. Six patients had a history of prolonged neonatal jaundice. Six patients died due to aspiration pneumonia and intractable seizure (Fig. 2 and Table 1).

a Presenting clinical features of included patients; b physical examination of studied patients
Presenting symptoms
The most common clinical features of our patients at first presentation was developmental delay (92%) with a mean age at presentation of 4.9 months, regression in speech with a mean age at presentation of 10.7 month and cognitive decline with a mean age at presentation of 9.39 months. Hypotonia (mean age at presentation 5.2 months), seizure (mean age at presentation 5.6 months), poor fix and follow and abnormal eye movement (mean age at presentation 2.7 months) were less common symptoms in order of frequency (Fig. 2 and Table 2).
Clinical findings
On physical examination organomegaly, especially splenomegaly was detected only in two patients (by physical examination and then ultrasonography). Organomegaly was not developed in other patients in 1 year follow-up. The age of the onset of organomegaly was uncertain and this sign found out during first examination and 1 year follow-up of the patients. Seven of cases had coarse facies. The majority of patients had truncal hypotonia (64%) and lower limbs spasticity (56%). On ophthalmologic examination, cherry red spots were detected in 17 patients (68%), the remaining patients did not show this sign. Diffuse Mongolian spots were seen in two patients and seven of them (28%) had hyperacusis as an important clue for clinical diagnosis of GM2 Gangliosidosis. Exaggerated patellar and ankle tendon reflexes were detected in 64% of the patients (Fig. 2). During the study, 11 infants including 6 male and 5 females died of aspiration pneumonia and intractable seizure.
Neuroimaging findings
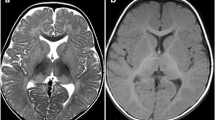
The most common findings in brain on MRI (Magnetic Resonance Imaging) that carried out for ten patients were hypomyelination and/or delayed myelination, supratentorial brain atrophy, abnormal signals in basal ganglia especially putamen, globus pallidus and caudate nucleus (Table 3). Five series of brain MRI showed a radiologic sign as titled due to bilateral thalamus marked hyperintensity in T1-weighted images (thalamus brightness) and marked hypointensity in T2-weighted images of brain MRI.
Laboratory findings and HEXB mutations
The activity of hexosaminidase A, B, AB was low in all of patients. In eight families studied by mutation analysis, seven disease-causing mutations were found. Two mutations have been previously reported (c.1538 T > C; p.Leu513Pro and c.850 > T; p.Arg284Ter) [6, 7]. Five mutations were novel; they comprised two missense mutations (c.1602C > A; p.Cys534Ter and c.833C > T; p.Ala278Val), a six bp tandem duplication (c.668_669 + 4dupTGGTAA) and a 16 bp frameshift deletion (c.1642_1657delGCTGGATATTGTAACC) (Table 4). All the parents were heterozygote for HEXB gene mutations.
Discussion
SD constitutes 7% of GM2 gangliosidosis with an incidence of 1 in 384,000 live births that is associated with decrease of β-hexosaminidase activity [1]. In this study, we reviewed the most common presenting symptoms, clinical course and outcome of 25 cases of infantile-onset Sandhoff disease. Although the clinical features of infantile SD are non-specific in most patients, our experience showed motor and cognitive milestones delay and regression as the most common and the earliest features of the disease [2, 8]. The mean age at presentation in our cases of 6.4 months and a delay of diagnosis of 7.8 months were similar to other reports [2, 9]. As we previously mentioned clinical symptoms of the infantile SD, organomegaly was detected in only 2 patients over the time and follow-up of the patients for at least 1 year, so, unlike other GM2 gangliosidosis the lack of organomegaly could not preclude the presence of infantile SD [8, 10, 11]. Cherry-red spots are a characteristic ocular and physical sign of the infantile SD and can be used for early detection of suspected patients to this disease [2, 8]. In present study as other reports association between diffuse Mongolian spots with some lysosomal storage diseases and SD has been showed that may be a clue for diagnosis of lysosomal storage disorders including infantile SD [12, 13]. Accumulation of calcium associated with collection of GM2 ganglioside leads to gliosis and loss of myelin and axon in the cortical neurons. These evolutions give rise some earliest findings on T2-weighted images of brain MRI including bilateral thalamic hypotensity and hypomylination that are characteristic of brain involvement in the infantile SD disease which also were detected in our patients [14, 15]. SD is caused by mutation in HEXB gene is about 40 kb in length and located on 5q13 with 14 exons [6]. So far, 51 mutations have been identified in HEXB, the most of which are missense/nonsense (the human gene mutation database). Although there is no correlation between genotype and phenotype in SD but knowledge about mutations will facilitate genetic counseling and prenatal diagnosis in affected families [16]. Genetic study is important to make definitive diagnosis and to help family planning and prenatal diagnosis in the affected families. The presence of various and novel mutations in our patients may indicate heterogeneity in mutations in infantile SD among the Iranian population [17, 18]. We expect these mutations lead to partial or complete loss of protein, because the activity of HEXB enzyme were lesser than normal.
Conclusion
Infantile SD should be considered for each child presented with neurologic regression especially during the first of life and presence of cherry red spots in ophthalmic examination. Absence of organomegaly cannot rule out the disease. Molecular analysis is important for definitive and prenatal diagnosis.
Abbreviations
- DBSs:
-
dried blood spots
- HEXB, A:
-
Hexosaminidase A, B
- INMR:
-
Neurometabolic registry
- ISD:
-
Infantile Sandhoff disease
- LSDs:
-
Lysosomal storage disorders
- MRI:
-
Magnetic resonance imaging
- PCR:
-
Polymerase chain reaction
Reference
Maegawa GHB, Stockley T, Tropak M, et al. The natural history of juvenile or subacute GM2 gangliosidosis: 21 new cases and literature review of 134 previously reported. Pediatrics. 2006;118(5):1550–62.
Smith NJ, Winston AM, Stellitano L, Cox TM, Verity CM. GM2 gangliosidosis in a UK study of children with progressive neurodegeneration: 73 cases reviewed. Developmental Medicine & Child Neurology. 2012;54:176–82.
Villamizar-Schiller IT, Pabón LA, Hufnage SB, Serrano NS, Karl G, John Jefferies JL, et al. Neurological and cardiac responses after treatment with miglustat and a ketogenic diet in a patient with Sandhoff disease. Eur J Med Genet. 2015;58(3):180–3.
Gomez-Lira M, Mottes M, Perusi C, Pignatti PF, Rizzuto N, Gatti R, Salviati A. A novel 4-bp deletion creates a premature stop codon and dramatically decreases HEXB mRNA levels in a severe case of Sandhoff disease. Mol Cell Probes. 2001;15:75–9.
Landegren U. Measurement of cell numbers by means of the endogenous enzyme hexosaminidase. Applications to detection of lymphokines and cell surface antigens. J Immunol Methods. 1984;67(2):379–88.
Zampieri S, Filocamo M, Buratti E, Stroppiano M, Vlahovicek K, Rosso N, et al. Molecular and functional analysis of the HEXB gene in Italian patients affected with Sandhoff disease: identification of six novel alleles. Neurogenetics. 2009;10:49–58.
Lee HF, Chi CS, Tsai CR. Early cardiac involvement in an infantile Sandhoff disease case with novel mutations. Brain and Development. 2017;39(2):171–6.
Karimzadeh P, Jafari N, Nejad Biglari H, Jabbeh Dari S, Ahmad Abadi F, Alaee MR, Nemati H, Saket S, et al. GM2-Gangliosidosis (Sandhoff and Tay Sachs disease): diagnosis and neuroimaging findings (an Iranian pediatric case series). Iran J Child Neurol. 2014;8(3):55–60.
Staretz-Chacham O, Lang TC, LaMarca ME, Krasnewich D, Sidransky E. Lysosomal storage disorders in the newborn. Pediatrics. 2009;123(4):1191–207.
Barness LA, Henry K, Kling P, Laxova R, Kaback M, Gilbert-Barness E. A 7-year old white-male boy with progressive neurological deterioration. Am J Med Genet. 1991;40(3):271–9.
Ozkara HA, Topcu M, Renda Y. Sandhoff disease in the Turkish population. Brain Dev. 1997;19(7):469–72.
Ashrafi MR, Tavasoli A, Shiva S, Parvaneh N, Tamizifar B. Diffuse dermal melanocytosis in two patients with Sandhoff disease and mucopolysaccharidosis VI. Int J Dermatol. 2014;53:736–8.
Ashrafi MR, Shabanian R, Mohammadi M, et al. Extensive Mongolian spots: a clinical sign merits special attention. Pediatr Neurol. 2006;34:143–5.
Lakshmi S, Fatima Shirly Anitha G, Vinoth S. A rare case of Sandhoff disease: two in the same family. Int J Contemp Pediatr. 2015;2(1):42–5.
Autti T, Joensuu R, Aberg L. Decreased T2 signal in the thalami may be a sign of lysosomal storage disease. Neuroradiology. 2007;49(7):571–8.
Zampieri S, Cattarossi S, Oller Ramirez AM, Rosano C, Lourenco CM, Passon N. Sequence and copy number analyses of HEXB gene in patients affected by Sandhoff disease: functional characterization of 9 novel sequence variants. PLoS One. 2012;7(7):e41516.
Gaignard P, Fagart J, Niemir N, Puech JP, Azouguene E, Dussau J, Caillaud C. Characterization of seven novel mutations on the HEXB gene in French Sandhoff patients. Gene. 2013;512(2):521–6.
Dastsooz H, Alipour M, Mohammadi S, Kamgarpour F, Dehghanian F, Fardaei M. Identification of mutations in HEXA and HEXB in Sandhoff and Tay-Sachs diseases: a new large deletion caused by Alu elements in HEXA. Human Genome Variation. 2018;5:18003.
Zampieri S, Filocamo M, Buratti E, Stroppiano M, Vlahovicek K, Rosso N, Bignulin E, Regis S, Carnevale F, Bembi B, Dardis A. Molecular and functional analysis of the HEXB gene in Italian patients affected with Sandhoff disease: identification of six novel alleles. neurogenetics. 2009;10(1):49–58.
Author information
Authors and Affiliations
Contributions
JZ carried out the molecular genetic studies, PR and AT in the design of the study, NP performed the statical analysis, ZR and MRA participated in coordination and helped to the draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tavasoli, A.R., Parvaneh, N., Ashrafi, M.R. et al. Clinical presentation and outcome in infantile Sandhoff disease: a case series of 25 patients from Iranian neurometabolic bioregistry with five novel mutations. Orphanet J Rare Dis 13, 130 (2018). https://doi.org/10.1186/s13023-018-0876-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-018-0876-5