
Abstract
Background
Alpha-1-antitrypsin (AAT) deficiency (AATD) of Z, Mmalton, Siiyama type is associated with liver storage of the mutant proteins and liver disease. The Z variant can be diagnosed on isoelectric focusing (IEF) while Mmalton and Siiyama may be missed or misdiagnosed with this technique. Therefore, molecular analysis is mandatory for their characterization. In particular, that holds true for the Mmalton variant as on IEF profile it resembles the wild M2 subtype.
Methods
This is a retrospective analysis involving review of medical records and of liver biopsy specimens from a series of Mmalton, Z and Siiyama Alpha-1-antitrypsin deficiency patients. The review has been implemented by additional histological stains, electron microscopic observations and 3-D modeling studies of the sites of the mutations.
Results
Z, Mmalton and Siiyama liver specimen contained characteristic intrahepatocytic PAS-D globules. The globules differed in the three variants as only Mmalton cases showed dark basophilic precipitates within the AAT inclusions. The precipitates were visualized in haematoxylin-eosin (H.E.) stained preparations and corresponded to calcium precipitates as demonstrated by von Kossa staining. On immunohistochemistry, ZAAT inclusions were stained by polyclonal as well as monoclonal noncommercial anti-AAT antibody (AZT11), whilst Mmalton and Siiyama inclusion bodies remained negative with the monoclonal anti-Z antibody. 3-D protein analysis allowed to predict more severe misfolding of the Mmalton molecule as compared to Z and Siiyama that could trigger anomalous interaction with endoplasmic reticulum chaperon proteins, namely calcium binding proteins.
Conclusions
Mmalton AAT inclusion bodies contain calcium precipitates inside them that allow the differential diagnosis with Siiyama and ZAAT inclusions in routine histological sections. The study has confirmed the specificity of the monoclonal AZT11 for the Z mutant. Thus, the combination of these two features is crucial for the distinction between the three variants and for predicting the genotype, whose confirmation would definitely require molecular analysis. Our study provides new data on the pathomorphogenesis of Mmalton inclusion bodies whose mineralization could play a central role in disease pathogenesis of Mmalton that is distinct from the Z and Siiyama variants. Calcium is known to be a major effector of cell death either via the increased intracellular concentration or the alteration of homeostasis.
Similar content being viewed by others


Background
Alpha-1-antitrypsin (AAT) deficiency (AATD), an autosomal co-dominant genetic disorder, was discovered in 1963 on the base of the recurrent lack of alpha-1-globulin electrophoretic peak in members of the same family [1]. Subsequently, an analogous electrophoretic pattern was observed in children with cryptogenic liver cirrhosis whose hepatocytes contained peculiar PAS diastase (PAS-D) inclusions which reacted positively with a polyclonal anti AAT antibody [2]. Later on it was demonstrated that the protein retained within the hepatocytes was affected by a mutation, i.e. the replacement of glutamic acid by lysine at position 342 (Glu342Lys) in exon V of the AAT gene [3]. On Isoelectric Focusing (IEF), the mutant protein showed a very slow migration and for that reason, it has been designated with the last letter of the alphabet, Z. This letter indicates the phenotype of the main variant of the protease inhibitor (PiZ). Following the demonstration that the mutant protein was retained within the endoplasmic reticulum as a consequence of an abnormal conformation of the molecule, AATD has become the prototype of a new group of diseases, the Endoplasmic Reticulum Storage Disease (ERSD) [4], designated also as Conformational Diseases (CD) [5]. Presently about 100 allelic variants of the AAT gene have been detected, a few of them having lower than normal serum levels in the absence of liver pathology. Only three variants, Z, Mmalton and Siiyama are characterized by liver storage of the protein and are at risk of developing chronic liver disease and/or pulmonary emphysema. Other very rare variants have been shown polymerization capability in cell models [6, 7] but so far there are no observations in liver tissue specimens of those variants. The S mutation, which is the most frequent deficiency variant, is never resulting in liver storage [8, 9] most probably because of the instability of the molecule and its degradation before secretion [10]. The unique variant PiNull has low or no circulating AAT, no liver disease but pulmonary emphysema [11]. The discovery of AATD has led to the understanding of the pathogenesis of pulmonary emphysema as due to the proteolytic action of neutrophilic elastases on lung elastic tissue because of the deficiency of AAT, the major protease inhibitor (Pi). In contrast, the pathogenesis of liver damage is not completely known. The most plausible hypothesis refers to the more pronounced apoptosis of hepatocytes carrying higher amount of insoluble polymerized/aggregated AAT within the Endoplasmic Reticulum (ER), whilst the soluble forms are degraded by proteasome [12].
The observation that not all AATD individuals with hepatic storage develop liver disease, has favored the hypothesis that additional factors either environmental or genetic could be involved. Genes that negatively regulate autophagy, thus increasing the hepatocytic apoptosis, are potential candidates.
In the clinical setting, the characterization of AAT is requested in the presence of low serum levels of the protein. The first level investigation is IEF that helps in establishing the phenotype in most cases.
Previous study have shown that the serum concentration determination can be unreliable in heterozygous PiMZ phenotype as these individuals, under conditions of clinical stimulation, are capable of rising their serum levels up to the normal range due to the acute phase reactant nature of the M component. This phenomenon has been called “Recuitment-Secretory Block” phenomenon [13].
In addition, IEF has some limitations: Mmalton individuals show a mobility pattern analogous to the normal M2 subtype allele. With Mmalton, the situation becomes more complex when the mutation is associated with Z mutation, as the latter per se can explain the IEF pattern as well as the low serum levels. Finally, the Mmalton protein in homozygous condition can be visualized on IEF but it is quite indistinguishable from the compound Null/Mmalton condition [14].
The second level diagnostic investigation is molecular genetics, i.e. the sequencing of the entire gene of AAT that defines the genotype and identifies the pathogenetic mutations.
Z, Mmalton and Siiyama AAT accumulates in parenchymal liver cells and is visualized under the light microscope in the form of PAS-D inclusions that are positively stained by anti-AAT polyclonal antibodies.
Up to now, the three variants cannot be distinguished on the basis of the sole morphology. Moreover, one cannot rely upon epidemiological data that indicate a very high incidence (up to 7% in the general population) in Northern Europe [15]. Mmalton is much less rare but still is the most frequent variant in Sardinia [16], an Italian island with a high incidence of AATD deficiency. Siiyama is very rare and apparently confined to Far East [17, 18].
With regard to the immunological properties of the three variants, previous studies have proven that a noncommercial monoclonal anti-ZAAT antibody (AZT11) recognizes exclusively and selectively only the Z variant [15, 19, 20]. Recently Joly et al. [14] have reported a positive staining of AAT inclusions in a Mmalton/Mmalton patient using a not otherwise specified monoclonal antibody to AAT.
In this paper, we report on novel morphological findings that can enable to distinguish the most severe AATD variants, i.e. Z, Mmalton and Siiyama, and to unravel the pathogenesis of the liver cell damage.
Methods
Study Groups
-
Group 1 (Mmalton patients)
Group 1 consisted of seven patients (Table 1), four adults and three children, undergoing a liver biopsy because of chronic elevation of liver enzymes and low plasma levels of AAT. The patients showed accumulation of AAT within hepatocytes and, on molecular analysis, displayed the Phe52deletion (Mmalton) either in heterozygous or in homozygous condition. In patient n.7 the liver biopsy had been performed after the genotyping, carried out first because of persistent very low serum AAT levels.
The main clinical-pathological features of Group 1 patients are summarized in Table 1.
-
Group 2 (Z patients)
Group 2 consisted of liver biopsies from seven patients (four adults and three children) diagnosed as Z AATD. Clinical-pathological features of Z patients are summarized in Table 2.
-
Group 3 (Siiyama patient)
Consisted of spared unstained histological sections from the Siiyama patient included in a previous study [19].
-
Group 4 (Controls)
Liver biopsy specimens from two normal MM AAT individuals were used as controls.
The liver biopsies from the four Groups were obtained retrospectively.
Morphological studies
Routine histology
Paraffin embedded liver tissue biopsy specimens were available from all Mmalton and Z AATD patients and from the Siiyama patient. The cases were selected on the base of the presence on routine histological examination, of eosinophilic PAS diastase resistant (PAS-D) inclusion bodies within hepatocytes.
Tissue sections were stained with hematoxylin-eosion (H.E.), PAS, PAS-D, Gomori’s reticulin, Masson’s trichrome, von Kossa for calcium and by immunohistochemistry with a polyclonal anti-AAT antibody. Additional serial sections were stained with a monoclonal antibody (AZT11) that recognizes specifically the Z variant as previously described [15, 19, 20].
Electron microscopy
A small fragment from needle or surgical specimens from all cases had been fixed in glutaraldheide and processed for transmission electron microscopy (TEM). Semithin sections were stained with toluidine blue. Ultrathin sections were mounted on copper grids, stained with lead citrate and uranyl acetate and examined with a Zeiss 109 TEM.
Molecular genetics analysis
DNA was isolated from total blood from all patients using extraction protocols from the QIAMP DNA mini kit (QIAGEN, Hilden, Germany), after the informed consent of patients or parents. Mutation screening was done using polymerase chain reaction (PCR), amplification and DNA sequencing of coding exons and all splices junction of AAT gene were carried out as previously described [20, 21].
Structural analysis
To represent the human AAT and to examine the amino acid environment around the sites affected by the Phe52deletion (Mmalton case) and Ser53Phe mutation (Siiyama), the Protein Data Bank (PDB) structure 1QLP was employed. The latent form of AAT was built by homology modelling using as the template the crystal structure of conserpin in the latent state (PDB entry 5CDZ). To display the Z form, the crystal structure of human AAT bearing the Glu342Lys amino acid change (PDB structure 5IO1) was employed.
The molecular volume of the crystal structure of the luminal domain of calnexin (PDB entry 1JHN) was used to infer the highest calcium concentration that can locally build up assuming tight aggregation of this protein and knowing that each molecule binds one Ca2+ ion.
Results
Morphological studies
Routine histology
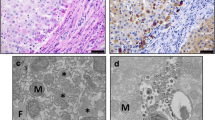
On routine histology Mmalton, Z and Siiyama cases showed a variable degree of morphological alterations ranging from mild chronic hepatitis to fully developed cirrhosis, two of which complicated by hepatocellular carcinoma. These two cases were already reported for purposes other than the present study [21]. On light microscopy all cases presented round eosinophilic inclusions within hepatocytes. The inclusions were strongly PAS-D positive. In six out of seven Mmalton patients a number of AAT inclusions were centered by dark basophilic precipitates whose sized ranged from small (Fig.1a) to very large inclusions within AAT globules (Fig.1b). The dark basophilic material within AAT inclusions was not visible in PAS-D preparations but was positively stained by von Kossa staining for calcium (Fig.1a , inset). The number of calcified inclusions was scored semiquantitatively: + corresponded to 1–5% of periportal hepatocytes, ++ to 6–15%, +++ to 16–30%. The liver biopsy from case n.7 (Table 1) consisted of a very small and fragmented needle specimen. In this case, very few AAT inclusions devoid of basophilic precipitates could be found. Calcium precipitates were not seen in any ZAAT nor in the Siiyama case. On immunostaining, the Mmalton and Siiyama inclusions were positively stained by the polyclonal AAT antibody but remained negative with the monoclonal anti-ZAAT antibody, whilst the Z inclusions were positively stained by both the polyclonal and the monoclonal ZAAT antibody as previously described [9, 19, 20]. No AAT inclusion bodies or calcium precipitates were observed in MM normal control cases.

(a) Liver tissue section from case 6 Table 1 (Mmalton/Mmalton), with a preserved lobular architecture. Hepatocytes contain eosinophilic inclusions identified as AAT. A few of them mostly located in check-border hepatocytes are centered by dark basophilic material corresponding to calcium precipitates, H.E. 40X, positively stained by von Kossa staining, 100X (Fig. 1a, inset). (b) Liver tissue section from case 1 Table 1 (Mmalton/Mmalton), with fully established cirrhosis, H.E. 40X. The microphotograph shows hepatocytes plenty of cytoplasmic eosinophilic inclusions that were PAS-D positive and immunoreactive with a polyclonal anti-AAT antibody. The largest AAT globule is centered by a dark basophilic material, positively stained by von Kossa staining for calcium
Electron microscopy
Under the EM, AAT material was localized in all cases within dilated cisternae of the ER. In six Mmalton cases, AAT inclusions displayed dark precipitates either in the form of granular non-crystalline or very electron dense crystalline material (Fig.2a). In the seventh Mmalton case (case n.7 Table 1) in which no calcium bodies were detected in H.E. stained preparations, fine electron dense granules quite similar to those of the other Mmalton cases were found in a few AAT inclusions (Fig.2b).

(a) Electromicrophotogaph from case 6 (Table 1) shows an hepatocyte with dilated cisternae of the ER containing fluffy semi-electron dense material corresponding to the classical appearance of AAT. The largest inclusions contain a large crystalline electrondense material. In addition to the crystalline structures, AAT inclusions contained fine or coarse electrondense granules quite similar to those of other AAT inclusions. This material was also displaying the peaks of calcium on EPMA (10.000 X). (b) Case 7 (a two years old boy with no visible calcium precipitates in H.E. stained preparations). The electronmicrophotograph shows dilated cisternae of ER. At least three AAT inclusions contain sparse electrondense granular precipitates similar to Fig. 2a. A few electrondense lysosomes with lipid inclusions are also seen (8000 X)
Structural analysis
AAT, as other serpin family members, presents an equilibrium between an active (Fig.3a) and latent (Fig.3b) form, with the latter derived from the former by relocation of the reactive center loop (RCL), which implies detachment of strand s1C from the β-sheet C region, and insertion of a portion of the RCL, structured as strand s4A, into the β-sheet A region between strands 3 and 5.

Sites of the Mmalton, Z and Siiyama mutations, and functional regions. (a) Active form of AAT structure (the β-strand region A is colored in blue). (b) Homology model of the latent form of AAT. (c) Superposition of the active AAT and Z variant structures. (d) Detailed view around the sites of Malton and Siiyama mutations highlighting surrounding core domain residues
The Z variant is the unique hepatocytic storage variant that has been structurally characterized, and its Glu342Lys change has been proposed to induce lability in strand s5A, which produces AAT intermediates with only a partial insertion of this strand into the central beta-sheet A yet in equilibrium with the inhibitory native-like fold [22]. In fact, the Z form has been solved in a conformation that essentially overlaps with that of the active form of the wild type AAT (Fig.3c).
Although the structure of the Mmalton protein is still unknown, dramatic conformational changes can be envisaged for this AAT variant since the Phe52deletion occurs beneath the beta-sheet A region and results in the “coalescence” of Phe51-Phe52 pair of residues into a single phenylalanine with the alteration of several intramolecular interactions crucial for the domain core stability (Fig.3d). Concerning the Siiyama mutation, it also involves a residue beneath the β-sheet A (Fig.3d), but in this case the change consists of the missense Ser53Phe replacement, which destabilizes the affected region but not as much as the Phe52deletion in Mmalton.
Discussion
Z, Mmalton and Siiyama deficiency variants of AAT are characterized by conformational abnormalities of amounts of the mutant molecules leading to insoluble polymerized/aggregated forms that accumulate in the ER of hepatocytes. Thus, the three variants fulfill the requirements for Endoplasmic Reticulum Storage Diseases (ERSD) [4] and Conformational Diseases [5].
AATD is worldwide distributed with a diverse incidence. The Z allele shows the highest incidence in the Northern Europe where it reaches about 7% of the general population [15]. The Siiyama is the most severe form of AATD in Far East [17, 18], Mmalton is the most common cause of AATD in the Italian island of Sardinia [16].
Individuals carrying the mutations present with reduced serum levels of AAT and accumulation of the mutant protein within hepatocytes.
The three forms are due respectively to the following mutations: Z to the missense Glu342Lys in exon V, Siiyama to the missense Ser53Phe in exon II and Mmalton to the Phe52deletion in exon II. The mutations are demonstrated by DNA sequencing of the entire AAT gene.
However, molecular genetics analysis is not available in all departments of Pathology, so that pathologists, in front of PAS-D hepatocytic inclusions, can only make the general diagnosis of AATD after proving that the stored material corresponds exclusively and selectively to AAT [4].
In this paper, we have provided a means to make feasible the differential diagnosis among the three variants in routine liver specimens. Indeed the three forms are indistinguishable on PAS-D staining and all react with polyclonal anti-AAT antibodies. However in contrast to the Z variant, Mmalton as well as Siiyama are not recognized by the monoclonal antibody (AZT11) that is specific for the Z variant [15, 19, 20]. Furthermore, only Mmalton presents with calcium precipitates visualized in H.E. stained preparations. This mineralization phenomenon does not occur with either Z or Siiyama AAT. Hence, it follows that the occurrence of calcium precipitates within AAT inclusions represents a diagnostic clue that per se would render unnecessary monoclonal antibody testing as well as molecular genetics analysis, especially if this technique is not available.
Molecular analysis remains the definite test also in Mmalton cases (like case n. 7 in our series) in which calcifications are not detected due to sampling error or insufficient tissue material.
To summarize, this study has unequivocally shown a striking difference between Mmalton, Z and Siiyama AATD in that Mmalton inclusions undergo a mineralization process and do not react with the monoclonal anti-ZAAT antibody. The two observations suggest the hypothesis that the two phenomena might have a relationship.
The monoclonal anti-ZAAT antibody appears to recognize a neoepitope lying close to position 342 and revealed upon formation of reactive loop β-sheet A polymers [19, 23, 24]. The negative staining of Mmalton inclusions with the Z antibody suggests that the structural perturbation leading to Mmalton polymers is different than that of the ZAAT polymerization. Indeed the Mmalton mutation has been shown to cause over insertion of the RCL and a likely C sheet mechanism of polymerization [23], whilst the Z mutation causes a partial insertion into β-sheet A, opening the lower part of β-sheet A, which then acts as an acceptor of the RCL of another ZAAT molecule [5, 23, 25].
In addition to definitely confirm in a large series of patients the impact of the Z mutation towards its immunological property, this study has led for the first time to the constant observation of calcium particles exclusively in samples from Mmalton patients, despite Z and Siiyama AAT presents similar fate of hepatocytic storage.
For the sake of completeness it has to be underlined that in our study we could examine a single Siiyama case and that one out of the seven reported Mmalton cases did not display calcifications on H.E. stained preparation.
However, to our knowledge, our Siiyama is the only one available case in the literature with liver histology. On the other side, the single Mmalton case from our study who did not show calcification on light microscopy, presented ultrastructural features quite analogous to the other six Mmalton cases. Once more, it is useful to emphasize that those features never occur in Z or Siiyama AATD variants. Of course, as calcifications of Mmalton globules represent a new observation, it would be important to check additional Mmalton livers as well as human liver tissue from other rare deficient variants capable of polymerization in vitro.
In order to explain the combination of the two phenomena observed in Mmalton, i.e. calcifications and lack of reactivity to the monoclonal ATZ11 antibody, we have inferred that Mmalton, among these three variants, is the most severely misfolded form of AAT and hypothesized that, in addition to hepatocytic storage, this mutant might also trigger damages by involving ER resident proteins. As a matter of facts, the ER functions as a storehouse of calcium ions, most of the resident proteins interact with AAT, and aberrant antitrypsin can activate endoplasmic reticulum-specific stress responses [26]. In this cellular defense mechanism, among the protein network evoked, calnexin, the major calcium transporter in the secretory pathway [27], which, in addition to binding calcium and regulating calcium homeostasis, has a molecular chaperoning function. Under stress, calnexin in C. elegans is present at higher levels and the overexpressed quote also binds calcium [28]. Furthermore, AAT is a well characterized cargo protein of calnexin [29], a physical association was seen between calnexin and a secretion-incompetent variant of human AAT [30], and calnexin was found to bind the alpha-1-antitrypsin null Hong Kong (NHK) releasing it, only once their interaction terminates, to the mammalian translocon-associated protein (TRAP) complex [31].
Concerning detrimental effects of AAT variants on the protein structure, it is worth to mention the work of Lomas et al. [23] who found that, compared to M or Z antitrypsin, the monomeric form of antitrypsin Mmalton had much slower rate in accepting an 11-mer antithrombin RCL peptide thus implying that this mutant harbors more important conformational changes in the A sheet region. The same authors also observed the unusual loss of the C-terminal peptide from the reactive loop-cleaved component of plasmatic antitrypsin, as this peptide normally remains tightly bound to the remainder of the domain.
On the other hand, biochemical indication that the defects of the Z form are much less severe and do not lead ineluctably to aggregations was provided by the evidences that the secretory impairment of Z antitrypsin can be corrected by chemical chaperons [32]. Furthermore, the insertion of the RCL peptide of one protein as strand s4A into the β-sheet A of another protein and so forth, proposed as mechanism of loop-sheet polymerization for aberrant antitrypsin forms, was successfully inhibited by introducing competitive exogenous RCL peptide fragments [33].
Finally, examination of the crystal structure of AAT in the active form shows that, among the Mmalton, Siiyama and Z mutation, the former (Phe52deletion) appears to be the most detrimental to structure integrity as it implies loss/alteration of several hydrophobic intramolecular interactions in the domain core beneath the β-sheet A region (Fig. 3).
This might seem at odds with the observation that the monomeric Mmalton apparently circulates in a normal functional form [23], but it is also true that antitrypsin inhibitory function relies on the flexible RCL peptide portion, which might remain available to its protease targets despite major rearrangements in the remainder of the AAT domain.
Conclusion
Based on these evidences, it can be thought that ER stress and the associated calnexin upregulation triggered by Mmalton may lead to unusually strong interactions with this chaperone preventing it to escape after failed attempts to recover the AAT fold. In this hypothesis, Mmalton-induced aggregation would trigger the accumulation of calnexin thus the building up of calcium pools initially in protein-ligated form. Eventually, with the aging of the protein aggregates, free Ca2+ ions can be released fostering calcification. Abnormal aggregation of calnexin molecules can be a factor producing saturating levels of calcium. In fact, by assuming tight associations of calnexin (and taking into account the molecular volumes inferred from the crystal structure of the luminal domain of calnexin), the resulting concentration of calcium would be ca. 0.05 mol/L. This value exceeds the concentration necessary for the precipitation of calcium salts with physiologically available anions.
We believe that further investigations, including functional studies, are required to fully clarify the intriguing phenomenon observed in this study, especially with regard to the possibility of calnexin’s involvement.
Anyhow, the results appear to be relevant in that the mineralization of AAT inclusion bodies occurs only with the Mmalton protein thus allowing the unequivocal differential diagnosis between Mmalton, Z and Siiyama AATD and could play a central role in disease pathogenesis that is distinct from the common Z variant. Liver cell necrosis has longly been associated with intracellular calcium accumulation or its homeostasis alteration [34].
We feel that our observation can be implemented to expand the analysis of conformational diseases, in terms of pathological burden, and we hope that further studies will provide more meaningful arguments for the relevance of the novel data. Likewise, it is hoped that further studies, either retrospective or prospective, could be performed on as many as possible Mmalton livers especially from areas like the Mediterranean one, where the mutation is not so uncommon.
References
Laurell CB, Eriksson S. The electrophoretic alpha1-globulin pattern of serum in alpha1-antitrypsin deficiency. 1963. Copd. 1963;10(Suppl 1):3–8.
Sharp HL, Bridges RA, Krivit W, Freier EF. Cirrhosis associated with alpha-1-antitrypsin deficiency: a previously unrecognized inherited disorder. J Lab Clin Med. 1969;73(6):934–9.
Owen MC, Carrell RW. alpha-1-Antitrypsin: sequence of the Z variant tryptic peptide. FEBS Lett. 1977;79(2):245–7.
Callea F, Brisigotti M, Fabbretti G, Bonino F, Desmet VJ. Hepatic endoplasmic reticulum storage diseases. Liver. 1992;12(6):357–62.
Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency--a model for conformational diseases. N Engl J Med. 2002;346(1):45–53.
Fra AM, Gooptu B, Ferrarotti I, Miranda E, Scabini R, Ronzoni R, Benini F, Corda L, Medicina D, Luisetti M, Schiaffonati L. Three new alpha1-antitrypsin deficiency variants help to define a C-terminal region regulating conformational change and polymerization. PLoS One. 2012;7(6):e38405.
Poller W, Merklein F, Schneider-Rasp S, Haack A, Fechner H, Wang H, Anagnostopoulos I, Weidinger S. Molecular characterisation of the defective alpha 1-antitrypsin alleles PI Mwurzburg (Pro369Ser), Mheerlen (Pro369Leu), and Q0lisbon (Thr68Ile). Eur J Hum Genet. 1999;7(3):321–31.
Callea F, Brisigotti M, Faa G, Lucini L, Eriksson S. Identification of PiZ gene products in liver tissue by a monoclonal antibody specific for the Z mutant of alpha 1-antitrypsin. J Hepatol. 1991;12(3):372–6.
Callea F, Fevery J, De Groote J, Desmet VJ. Detection of Pi Z phenotype individuals by alpha-1-antitrypsin (AAT) immunohistochemistry in paraffin-embedded liver tissue specimens. J Hepatol. 1986;2(3):389–401.
Carrell RW, Jeppsson JO, Laurell CB, Brennan SO, Owen MC, Vaughan L, Boswell DR. Structure and variation of human alpha 1-antitrypsin. Nature. 1982;298(5872):329–34.
Cox DW, Levison H. Emphysema of early onset associated with a complete deficiency of alpha-1-antitrypsin (null homozygotes). Am Rev Respir Dis. 1988;137(2):371–5.
Perlmutter DH. Autophagic disposal of the aggregation-prone protein that causes liver inflammation and carcinogenesis in alpha-1-antitrypsin deficiency. Cell Death Differ. 2009;16(1):39–45.
Callea F, Fevery J, Massi G, Lievens C, de Groote J, Desmet VJ. Alpha-1-antitrypsin (AAT) and its stimulation in the liver of PiMZ phenotype individuals. A "recruitment-secretory block" ("R-SB") phenomenon. Liver. 1984;4(5):325–37.
Joly P, Guillaud O, Hervieu V, Francina A, Mornex JF, Chapuis-Cellier C. Clinical heterogeneity and potential high pathogenicity of the Mmalton Alpha 1 antitrypsin allele at the homozygous, compound heterozygous and heterozygous states. Orphanet J Rare Dis. 2015;10:130.
Wallmark A, Alm R, Eriksson S. Monoclonal antibody specific for the mutant PiZ alpha 1-antitrypsin and its application in an ELISA procedure for identification of PiZ gene carriers. Proc Natl Acad Sci U S A. 1984;81(18):5690–3.
Ferrarotti I, Baccheschi J, Zorzetto M, Tinelli C, Corda L, Balbi B, Campo I, Pozzi E, Faa G, Coni P, Massi G, Stella G, Luisetti M. Prevalence and phenotype of subjects carrying rare variants in the Italian registry for alpha1-antitrypsin deficiency. J Med Genet. 2005;42(3):282–7.
Ko DH, Chang HE, Song SH, Yoon H, Park KU, Song J. Identification of compound heterozygous mutation in a Korean patient with alpha 1-antitrypsin deficiency. Korean J Lab Med. 2011;31(4):294–7.
Seyama K, Hirai T, Mishima M, Tatsumi K, Nishimura M. Respiratory Failure Research Group of the Japanese Ministry of Health, L.; Welfare, A nationwide epidemiological survey of alpha1-antitrypsin deficiency in Japan. Respir Investig. 2016;54(3):201–6.
Janciauskiene S, Eriksson S, Callea F, Mallya M, Zhou A, Seyama K, Hata S, Lomas DA. Differential detection of PAS-positive inclusions formed by the Z, Siiyama, and Mmalton variants of alpha1-antitrypsin. Hepatology. 2004;40(5):1203–10.
Sergi C, Consalez GG, Fabbretti G, Brisigotti M, Faa G, Costa V, Romeo G, Callea F. Immunohistochemical and genetic characterization of the M Cagliari alpha-1-antitrypsin molecule (M-like alpha-1-antitrypsin deficiency). Lab Invest. 1994;70(1):130–3.
Francalanci P, Santorelli FM, Saccani S, Bonetti MF, Medicina D, Coni P, Faa G, Callea F. Z and Mmalton-1-antitrypsin deficiency-associated hepatocellular carcinoma: a genetic study. Liver Int. 2009;29(10):1593–6.
Huang X, Zheng Y, Zhang F, Wei Z, Wang Y, Carrell RW, Read RJ, Chen GQ, Zhou A. Molecular Mechanism of Z alpha1-Antitrypsin Deficiency. J Biol Chem. 2016;291(30):15674–86.
Lomas DA, Elliott PR, Sidhar SK, Foreman RC, Finch JT, Cox DW, Whisstock JC, Carrell RW. alpha 1-Antitrypsin Mmalton (Phe52-deleted) forms loop-sheet polymers in vivo. Evidence for the C sheet mechanism of polymerization. J Biol Chem. 1995;270(28):16864–70.
Lomas DA, Mahadeva R. Alpha1-antitrypsin polymerization and the serpinopathies: pathobiology and prospects for therapy. J Clin Invest. 2002;110(11):1585–90.
Elliott PR, Lomas DA, Carrell RW, Abrahams JP. Inhibitory conformation of the reactive loop of alpha 1-antitrypsin. Nat Struct Biol. 1996;3(8):676–81.
Lawless MW, Greene CM, Mulgrew A, Taggart CC, O'Neill SJ, McElvaney NG. Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z alpha 1-antitrypsin deficiency. J Immunol. 2004;172(9):5722–6.
Ou WJ, Cameron PH, Thomas DY, Bergeron JJ. Association of folding intermediates of glycoproteins with calnexin during protein maturation. Nature. 1993;364(6440):771–6.
Lee W, Lee TH, Park BJ, Chang JW, Yu JR, Koo HS, Park H, Yoo YJ, Ahnn J. Caenorhabditis elegans calnexin is N-glycosylated and required for stress response. Biochem Biophys Res Commun. 2005;338(2):1018–30.
Chevet E, Jakob CA, Thomas DY, Bergeron JJ. Calnexin family members as modulators of genetic diseases. Semin Cell Dev Biol. 1999;10(5):473–80.
Le A, Steiner JL, Ferrell GA, Shaker JC, Sifers RN. Association between calnexin and a secretion-incompetent variant of human alpha 1-antitrypsin. J Biol Chem. 1994;269(10):7514–9.
Nagasawa K, Higashi T, Hosokawa N, Kaufman RJ, Nagata K. Simultaneous induction of the four subunits of the TRAP complex by ER stress accelerates ER degradation. EMBO Rep. 2007;8(5):483–9.
Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc Natl Acad Sci U S A. 2000;97(4):1796–801.
Alam S, Wang J, Janciauskiene S, Mahadeva R. Preventing and reversing the cellular consequences of Z alpha-1 antitrypsin accumulation by targeting s4A. J Hepatol. 2012;57(1):116–24.
Orrenius S, Mc Conkey DJ, Nicotera P. Disruption of intacellular Ca2+ homeostasis during oxidative stress. In: Heilmann C, editor. Calcium-Dependent Processes in the Liver. Lancaster: MTP press limited, Kluver Accademic Publishers Group; 1988. p. 185–6.
Acknowledgements
The authors wish to thank Marta Stefanelli, Federica Salutari and Riccardo Mariani for expert technical assistance, Mr. Gabriele Bacile for preparing the photographic material and Mrs. Nuccia Gatta, President of the Italian Association for Alpha-1-antitrypsin deficiency, for her enthusiastic encouragement.
Ethical approval and consent to participate
The study was approved by the Institutional Scientific Board of the Bambino Gesù Children’s Hospital and written informed consent was obtained from all patients.
Availability of data and materials
All data generated and analyzed materials during this study are included in this published article.
Author information
Authors and Affiliations
Author notes
Prof. Kamal Ishak, Head of the AFIP Washington, has deceased in 2004. Dr. Ishak has contributed with one case from AFIP files.
- Kamal Ishak
Contributions
FC and EB have planned the work described in the article and prepared the manuscript. RB has planned the EM study. GF has provided two cases, KS has provided the Syiiama case. VJD has provided one case. VN has collected the clinical data and the blood collection from three Mmalton children. IG e DM have conducted molecular analysis; EB has conducted homology modeling analysis; PF and FC analyzed histological studies. KI has contributed with one case with full investigations. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Consent for publication
The patients or their parents/guardians gave a written consent to publication of their anonymized clinical data.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Callea, F., Giovannoni, I., Francalanci, P. et al. Mineralization of alpha-1-antitrypsin inclusion bodies in Mmalton alpha-1-antitrypsin deficiency. Orphanet J Rare Dis 13, 79 (2018). https://doi.org/10.1186/s13023-018-0821-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-018-0821-7