
Abstract
Background
Histiocytic sarcoma (HS) is an aggressive malignant neoplasm. HS in the central nervous system is exceptionally rare and associated with a poor prognosis. This report documents a case of primary HS of the central nervous system with treatment including surgery, radiotherapy, and chemotherapy.
Case presentation
Our patient was a 47 year old female presenting with progressive ataxia, headaches, imbalance, nausea, vomiting, and diplopia. MRI showed a heterogeneously enhancing lesion approximately 2.9 × 3.0 × 2.3 cm centered upon the cerebellar vermis with mild surrounding vasogenic edema and abnormal enhancement of multiple cranial nerves. The patient underwent surgical debulking, which revealed histiocytic sarcoma with grossly purulent drainage. Staging revealed diffuse leptomeningeal involvement, primarily involving the brain and lower thoracic and lumbar spine. She underwent adjuvant radiotherapy to the brain and lower spine and was started on high dose methotrexate. However, she experienced progressive disease in the cervical and thoracic spine as well as pulmonary involvement. Genomic sequencing of her tumor showed a mutation in the platelet-derived growth factor receptor A (p.V0681) which could be targeted with Dasatinib. However, she did not tolerate Dasatinib and she succumbed to progressive disseminated disease eight months from original diagnosis. Our pathologic evaluation also revealed expression of PD-L1 and PD-L2 by tumor cells raising the potential therapeutic role for immune checkpoint inhibition.
Conclusions
This case provides an example of effective CNS control with resection and moderate doses of radiation therapy. A review of the literature confirms aggressive multidisciplinary treatment is the most effective treatment against this disease. In addition, genomic sequencing may play an important role in determining new therapeutic options. However, CNS histiocytic sarcoma remains an aggressive disease with a propensity for early widespread dissemination and few long term survivors.
Similar content being viewed by others


Background
Histiocytic sarcoma (HS) is an uncommon malignant neoplasm with morphological and immunophenotypic features of histiocytic tissue. HS presents most commonly with a mass involving soft tissues, gastrointestinal tract, and/or lymph nodes [16]. Primary histiocytic sarcoma of the central nervous system (CNS) is extremely rare and accounts for less than 1% of all lymphohematopoietic neoplasms [22]. A typical finding with CNS HS is the presence of an inflammatory infiltrate and the abundance of inflammatory cells can cause neoplastic cells to be overlooked and thus a misdiagnosis may occur [2]. Many previous cases are thought to be misdiagnosed due to lack of genetic markers, but the disease can now be recognized by its biological markers, such as positive expression of CD 163, CD 68, and lysozyme, which allow differentiation between CNS HS and other hematopoietic neoplasms [5], such as B-cell or T-cell non-Hodgkin’s Lymphoma [16, 21]. While the optimal treatment for CNS HS is not well defined, surgery is the primary modality with radiotherapy and chemotherapy frequently used as adjuvant treatments. Overall, CNS HS carries a poor prognosis, but there have been reported cases of long term disease free survival in adults when multidisciplinary treatment of radiation therapy, chemotherapy, and surgery are applied [2, 11, 23]. Here, we present a case of primary CNS HS with leptomeningeal spread treated with tri-modality therapy and provide a review of the available literature for this aggressive disease.
Case presentation
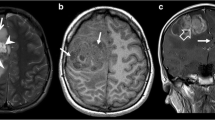
Our patient was a 47 year old female with a history of ulcerative colitis, Sjogren’s syndrome, migraines, and fibromyalgia who presented with a 6 month history of left lower extremity paresthesia followed by a 2 month history of progressive headaches, imbalance, ataxia, nausea, vomiting, and diplopia. Neurologic examination revealed severe gait ataxia requiring assistance to stand or walk and nystagmus with lateral gaze. MRI revealed a 3.0 cm heterogeneously enhancing mixed cystic and solid mass centered upon the cerebellar vermis with mild surrounding vasogenic edema and abnormal thickened enhancement of the bilateral vestibular nerves, left facial nerve, and right trigeminal nerve (Fig. 1).

a) Axial T1 Post Gadolinium Fat Saturation. b) T2 FLAIR Fat Saturation
The patient was promptly started on steroids, admitted to the hospital, and underwent a midline suboccipital craniotomy for debulking of the large intracerebellar tumor. Upon entering the cystic cavity, grossly purulent material without hemorrhage was noted. Gram stain and cultures were negative for infection. A subtotal resection was achieved and final pathology rendered the diagnosis of primary CNS histiocytic sarcoma (see Fig. 2). The H&E section (Fig. 2a and b) revealed sheets of large neoplastic cells with marked cytological atypia, brisk mitosis with occasional multilobated nuclei, and focal necrosis. Extensive immunohistochemical studies (Fig. 2c and d) showed the neoplastic cells positive for CD163, CD68, CD45, and Vimentin; negative for CD20, CD3, CD30, s-100, CD1a, CD21, CD23, pancytokeratin, MPO, CD61, CD123, GFAP, and BRAF. Further immunostains for PD-L1 with two different antibody clones (22C3 and 28–8) were also performed and showed more than 50% of the tumor cells were positive with membrane stain (Fig. 2e and f).

Final Pathological Examination. The H&E section show the diffuse proliferation of neoplastic histiocytes invading normal brain parenchyma (a × 10, upper normal brain; bottom neoplastic histiocytes); the neoplastic histiocytes show marked cytological atypia with brisk mitosis (b × 20). Immunohistochemical studies show the histiocytes positive for CD 163 (c ×10), CD68 (d × 10), CD45 and vimentin; negative for CD20, CD3, CD30, s-100, CD1a, CD21, CD23, pancytokeratin, MPO, CD61, CD123, GFAP, and BRAF. The neoplastic histiocytes are also positive for PD-L1 with both clones (e× 10, 22C3 and f× 10, 28–8)
Post-operative MRI showed a midline posterior occipital craniotomy with subtotal resection of tumor and continued bilateral-enhancement along multiple cranial nerves, concerning for leptomeningeal spread (Fig. 3).

a) Axial T1 Post Gadolinium Fat Saturation. b) T2 FLAIR Fat Saturation
A staging PET scan was performed two weeks following surgery suggesting diffuse leptomeningeal spread. A complete spinal MRI confirmed diffuse leptomeningeal spread in the lower thoracic and lumbar spine as well as cauda equina involvement. Due to the convincing evidence on MRI, a CSF analysis was deferred. Standard lab work was within limits, including LDH (Fig. 4).

a) Sagittal T2. b) Sagittal T1 Post Contrast
Three weeks following surgery, due to progressive lower back pain, she initiated radiation therapy including whole brain radiotherapy (30.6 Gy in 17 fractions) followed by a posterior fossa boost (5.4 Gy in 3 fractions) and a gross tumor boost (9 Gy in 5 fractions), for a total dose of 45 Gy in 25 fractions to gross disease with simultaneous radiation therapy to the lower thoracic and lumbar spine, receiving 40 Gy in 20 fractions, encompassing the areas of leptomeningeal disease in the spine and cauda equina. Full craniospinal radiotherapy was not performed to limit profound cytopenias that may have prevented further cytotoxic chemotherapy.
Interval follow up MRI showed a partial response with persistent posterior fossa disease but near complete resolution of previous leptomeningeal enhancement. Our plan was to initiate chemotherapy with a primary CNS lymphoma regimen with high CNS penetrating therapeutic agents including high-dose methotrexate, high-dose Ara-C, and thiotepa followed by high-dose chemotherapy with BCNU and thiotepa rescued by autologous stem cell transplantation. The patient was started on high dose intravenous methotrexate (HD-MTX) therapy, but received only one cycle secondary to patient intolerance.
A CT of the chest approximately two weeks after initiation of HD-MTX therapy for left sided rib pain showed new pulmonary nodules and a left sided pleural effusion, suspicious for metastatic disease. Thoracentesis revealed findings consistent with a malignant pleural effusion. The patient was initiated on cladribine (Leustatin).
Within two weeks after the completion of this infusion, the patient developed headaches, somnolence, fever, and nuchal rigidity. Malignant meningitis was suspected and the patient was started on IV antibiotics. Lumbar puncture was recommended but the patient declined. Her mental status significantly improved following antibiotic treatment. Brain MRI showed continued response of the cerebellar residual disease and no new intra-cranial lesions, but an MRI of the cervical spine showed new lesions consistent with metastatic disease in the untreated cervical spinal cord and thoracic vertebrae, including intramedullary involvement. She underwent further radiation therapy targeting C4-C6 and T1-T7 (20 Gy in 5 fractions). The patient rapidly showed improvement in symptoms.
Genomic sequencing of her tumor showed a novel mutation in the platelet-derived growth factor receptor A (p.V608I) and therefore, she was started on Dasatinib, a tyrosine kinase inhibitor (TKI) with known CNS penetration [24]. Unfortunately, this medication was poorly tolerated because of nausea, diarrhea, and acute pancreatitis so it was discontinued after one week.
The patient was readmitted to the hospital for continued decline in functional status, weakness, and failure to thrive. CT images of the chest, abdomen, and pelvis showed progressive bilateral pulmonary nodules. An MRI of the spine showed persistent but significantly improved leptomeningeal enhancement. Due to progression of symptoms, worsening systemic disease despite control of CNS disease, and limited systemic options available, the patient chose to enroll in hospice. The patient expired four weeks later, eight months after initial diagnosis.
Discussion
Histiocytic sarcoma of the CNS is an uncommon diagnosis with only about 30 total cases reported in the literature. The median age for diagnosis of HS is 46 years, which is equally distributed in males and females [20]. The prognosis for HS of the CNS is extremely poor with a median survival of 4.5 months, the longest recurrence free period reported being 42 months [6, 23].
This case was unique and merits discussion for several reasons. First, at the time of surgery, entry into the cerebellar lesion was associated with drainage of purulent material, which has been documented previously in only three other cases [1, 3, 26]. This was an unexpected finding at the time of surgery and no evidence of infection was identified. Second, this patient had extensive and symptomatic leptomeningeal disease with cranial nerve and spinal involvement by MRI. Leptomeningeal disease is relatively common in CNS HS, 10/31 (32%) documented cases had leptomeningeal involvement, which is associated with a poor quality of life and poor survival. In our case, this disease was treated aggressively with moderate dose radiation therapy, 45 Gy to the gross tumor in the cerebellum and 40 Gy to the leptomeningeal disease in the spine, which did achieve continued disease control, even 6 months after treatment. This raises the question as to whether patients with leptomeningeal spread would benefit from craniospinal radiotherapy. However, full craniospinal radiotherapy can induce profound cytopenias that could limit or delay the use of systemic therapy which has known benefit for histiocytic sarcoma. The other important aspect of this case is that we were able to obtain adequate amount of tissue from surgical resection of the brain tumor and performed genomic sequencing revealing a novel PDGFR mutation which is highly targetable with TKI’s. There is very limited literature regarding genomic data in CNS histiocytic sarcoma. One additional case report describes a patient with CNS histiocytic sarcoma with genomic sequencing revealing BRAF V600E mutation. The patient experienced a dramatic clinical and radiographic response with vemurafenib, though the response was not durable and the patient expired 6 months after initiation of treatment [17]. Our pathologic evaluation showed PD-L1 and PD-L2 expression by the tumor, which suggests that immunotherapy with checkpoint inhibitors should be explored. Using inhibitors to suppress PD-L1/PD-1 interaction has shown promising effects for treatment of various advanced cancers, but no literature is available for HS of the CNS [12]. Overexpression of PD-1 in an animal model has suggested antitumor immunity may be suppressed in histiocytic sarcoma, although this is yet to be shown in humans [27]. To the best of our knowledge, our case is the first report of PD-L1/PD-L2 expression in histiocytic sarcoma of the CNS.
A review of the literature for CNS HS was performed and the identified cases are shown in Table 1. Only three of thirty patients (10%) were alive with no evidence of disease at the time of publication, 16, 23, and 42 months after presentation [2, 11, 23]. Treatment for these cases consisted of surgery followed by concurrent chemoradiation with temozolomide and high dose focal radiation. In the two cases with radiation dose reported, the total dose given was 54 Gy and 61.4 Gy. This is consistent with the findings in our case that moderate doses of radiation are needed for local control.
The majority of cases in the literature received some form of surgery, radiation, and/or chemotherapy. The current standard treatment for CNS HS is surgery to achieve a maximal safe resection and for tissue diagnosis and relieving cerebral edema. An aggressive, multimodality approach is preferred with postoperative radiation therapy +/− chemotherapy, when the disease is unifocal. When the disease has become multifocal, further aggressive radiotherapy and combination chemotherapy is essential. Chemotherapeutics used previously include temozolomide (TMZ), cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP), rituximab, ifosfamide, carboplatin, etoposide (ICE), methotrexate, and thalidomide. There are no documented cases in the literature of successful treatment of multifocal disease. Thus, HS is a very aggressive disease when found in the central nervous system which warrants aggressive treatment with multiple modalities. Despite limited long term survivals of this disease, there have been approximately 10% of cases with long term survival with aggressive treatment including surgery, dose escalated radiation therapy, and chemotherapy.
Conclusion
In this case we present a patient with a primary histiocytic sarcoma of the CNS, an extremely rare diagnosis with a poor prognosis. This patient was treated aggressively with surgery, radiation therapy, chemotherapy, and targeted therapy informed by genomic sequencing but ultimately expired of her progressive disease approximately 8 months after diagnosis. The genomic sequencing of the tumor tissue showed a novel PDGFR mutation, which could be targeted with TKI’s. We showed expression of PD-L1 and PD-L2 by the tumor cells suggesting potential therapeutic role for immune checkpoint inhibitors. Our case demonstrates a durable response of CNS disease with radiation doses 40 Gy and above. Therefore, we recommend aggressive treatment with curative radiation doses to the CNS whenever appropriate and possible. Novel therapeutic agents such as TKI’s and checkpoint inhibitors need to be explored.
Abbreviations
- BCNU:
-
Carmustine
- CD:
-
Cluster of differentiation
- CHOP:
-
Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone
- CNS:
-
Central Nervous System
- CSF:
-
Cerebrospinal Fluid
- CT:
-
Computed Tomography
- GFAP:
-
Glial fibrillary acidic protein
- Gy:
-
Grey
- H&E:
-
Haemotoxylin and Eosin
- HD-MTX:
-
High Dose Methotrexate
- HS:
-
Histiocytic Sarcoma
- ICE:
-
Ifosfamide, Carboplatin, and Etoposide
- IV:
-
Intravenous
- LDH:
-
Lactate Dehydrogenase
- MPO:
-
Myeloperoxidase
- MRI:
-
Magnetic Resonance Imaging
- PD-1:
-
Programmed Cell Death Protein 1
- PDGFR:
-
Platelet Derived Growth Factor Receptor
- PD-L1:
-
Programmed Death-Ligand 1
- PD-L2:
-
Programmed Death Ligand-2
- PET:
-
Positron Emission Tomography
- TKI:
-
Tyrosine Kinase Inhibitor
- TMZ:
-
Temozolomide
References
Almefty RO, Tyree TL, Fusco DJ, Coons SW, Nakaji P. Primary histiocytic sarcoma of the brain mimicking cerebral abscess. J Neurosurg Pediatr. 2013;12:251–7.
Bai J, Li G, Shen M, Sui D, Lin S. Primary central nervous system histiocytic sarcoma mimicking glioma. Neurol India. 2014;62(6):684–5.
Bell SL, Hanzely Z, Alakandy LM, Jackson R, Stewart W. Primary meningeal histiocytic sarcoma: a report of two unusual cases. Neuropathol Appl Neurobiol. 2012;38:111–4.
Brown AF, Fan H, Floyd JR, Henry JM, Higgins RA. Primary central nervous system histiocytic sarcoma arising after precursor B-cell acute lymphoblastic leukemia. J Neuropathol Exp Neurol. 2015;74(12):1120–6.
Campo E, Swerdlow SH, Harris NL, et al. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117:5019–32.
Cao M, Eshoa C, Schultz C, Black J, Zu Y, Chang CC. Primary central nervous system histiocytic sarcoma with relapse to mediastinum: a case report and review of the literature. Arch Pathol Lab Med. 2007;131:301–5.
Chalasani S, Hennick M, Hocking WG, Shaw GR, Lawler B. Unusual presentation of a rare cancer: histiocytic sarcoma in the brain 16 years after treatment for acute lymphoblastic leukemia. Clin Med Res. 2013;11(1):31–5.
Chen CJ, Williams E, McAneney TE, Williams BJ, Mandell JW, Shaffrey ME. Histiocytic sarcoma of the cavernous sinus: case report and literature review. Brain Tumor Pathol. 2015;32(1):66–71.
Cheuk W, Walford N, Lou J, Lee AK, Fung CF, Au KH, et al. Primary histiocytic lymphoma of the central nervous system: a neoplasm frequently overshadowed by a prominent inflammatory component. Am J Surg Pathol. 2001;25:1372–9.
Devic P, Androdias-Condemine G, Streichenberger N, Berger F, Honnorat J, Broussolle E, et al. Histiocytic sarcoma of the central nervous system: a challenging diagnosis. QJM. 2012;105:77–9.
Foster M, Kamaly-Asl I, Stivaros S, Kelsey A, Gattamenini R, Kilday JP. Primary cerebral histiocytic sarcoma in childhood: a case report of protracted survival and review of the literature. Childs Nerv Syst. 2015;31(12):2363–8.
Gatalica Z, Bilalovic N, Palazzo JP, et al. Disseminated histiocytoses biomarkers beyond BRAFV600E: frequent expression of PD-L1. Oncotarget. 2015;6(23):19819–25.
Gentzler RD, Kahn D. Histiocytic sarcoma: a case of a 52-year-old female with two synchronous primary malignancies at presentation. Medicine Forum. 2011;11:31–3.
Gill-Samra S, Ng T, Dexter M, Wong M, Nahar N, Allsopp K, et al. Histiocytic sarcoma of the brain. J Clin Neurosci. 2012;19:1456–8.
Gomi K, Tanaka M, Yoshida M, Ito S, Sonoda M, Iwasaki F, et al. Primary cerebellar histiocytic sarcoma in a 17-month-old girl. J Neurosurg Pediatr. 2012;10:126–9.
Hornick JL, Jaffe ES, Fletcher CDM. Extranodal histiocytic sarcoma—clinicopathologic analysis of 14 cases of a rare epithelioid malignancy. Am J Surg Pathol. 2004;28:1133–44. 2004
Idbaih A, Mokhtari K, Emile JF, et al. Dramatic response of a BRAF V600E-mutated primary CNS histiocytic sarcoma to vemurafenib. Neurology. 2014;83(16):1478–80.
Kim YH, Yie GH, Kim NR, Jeon I, Cho HY, Seok JY, Kim EY, Lee KC. Pediatric intracerebral histiocytic sarcoma with rhabdoid features: case report and literature review. Neuropathology. 2017;37(6):560–8.
Laviv Y, Zagzag D, Fichman-Horn S, Michowitz S. Primary central nervous system histiocytic sarcoma. Brain Tumor Pathol. 2013;30:192–5.
Machado ES, de Miranda AC, Escopelli T, Caron R, Escopelli AC. Histiocytic sarcoma. Rev Bras Hematol Hemoter. 2011;33(2):155–7. https://doi.org/10.5581/1516-8484.20110038.
Marguet F, Piton N, Adle-Biassette H, Renaud F, Bohers E, Boyer T, Zarea A, Derrey S, Sabourin JC, Laquerriere A. Molecular characteristics of multifocal brain histiocytic sarcoma. Neuropathol Appl Neurobiol. 2018; https://doi.org/10.1111/nan.12490.
Moulignier A, Mikol J, Heran F, Galicier L. Isolated III cranial nerve palsies may point to primary histiocytic sarcoma. BMJ Case Rep. 2014; https://doi.org/10.1136/bcr-2014-204663.
Pérez-Ruiz E, Delgado M, Sanz A, Gil AM, Domínguez AR. Primary leptomeningeal histiocytic sarcoma in a patient with a good outcome: a case report and review of the literature. J Med Case Rep. 2013;13(7):127.
Porkka K, Koskenvesa P, Lundan T, et al. Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood. 2008;112(4):1005–12.
So H, Kim S, Yoon DH, et al. Primary histiocytic sarcoma of the central nervous system. Cancer Res Treat. 2015;47(2):322–8.
Sun W, Nordberg ML, Fowler MR. Histiocytic sarcoma involving the central nervous system: clinical, immunohistochemical, and molecular genetic studies of a case with review of the literature. Am J Surg Pathol. 2003;27:258–65.
Tagawa M, Maekawa N, Konnai S, Takagi S. Evaluation of costimulatory molecules in peripheral blood lymphocytes of canine patients with histiocytic sarcoma. PLoS One. 2016;11(2):e0150030. https://doi.org/10.1371/journal.pone.0150030. eCollection 2016
Torres CF, Korones DN, Powers JM, Vadasz AG. Primary leptomeningeal histiocytic lymphoma in a young child. Med Pediatr Oncol. 1996;27:547–50.
Toshkezi G, Edalat F, O'Hara C, Delalle I, Chin LS. Primary intramedullary histiocytic sarcoma. World Neurosurg. 2010;74:523–7.
Ueno T, Nishijima H, Kurotaki H, Kurose A, Tomiyama M. An unusual case of chronic meningitis due to histiocytic sarcoma of the central nervous system with meningeal dissemination. Neurol Sci. 2016;37(11):1875–7.
Wang J, Li T, Chen H, Liu Q. A case of primary central nervous system histiocytic sarcoma. Clin Neurol Neurosurg. 2012;114:1074–6.
Wu W, Tanrivermis Sayit A, Vinters HV, Pope W, Mirsadraei L, Said J. Primary central nervous system histiocytic sarcoma presenting as a postradiation sarcoma: case report and literature review. Hum Pathol. 2013;44:1177–83.
Zanelli M, Ragazzi M, Marchetti G, Bisagni A, Principi M, Fanni D, Froio E, Serra S, Zanetti E, De Marco L, Giangaspero F, Ascani S. Primary histiocytic sarcoma presenting as diffuse leptomeningeal disease: case description and review of the literature. Neuropathology. 2017; https://doi.org/10.1111/neup.12390,%202017.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Author information
Authors and Affiliations
Contributions
JMM Collected patient data, composed manuscript, and produced final draft. MRW Assisted in composing drafts of manuscript. WCS Provided revisions for manuscript. TAK Provided edits for manuscript. DHM Assisted with manuscript composition. BCM Assisted with manuscript composition and provided recommendations to article. RCM Provided initial concept for research and revised manuscript. LJ Provided pathology report and excerpt regarding markers included in manuscript. GWS Collected patient’s information including physician notes, procedure notes, and MRIs. DMT He revised and provided recommendations for the manuscript. KLC He provided edits and recommendations for the manuscript. RR Treated the patient and reviewed the manuscript. HWT Treated the patient and provided edits and recommendations for manuscript. JLP Treated the patient and provided major edits to manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This research was deemed ethical by our institution’s IRB review policy.
Consent for publication
Verbal consent was given by the patient.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
May, J.M., Waddle, M.R., Miller, D.H. et al. Primary histiocytic sarcoma of the central nervous system: a case report with platelet derived growth factor receptor mutation and PD-L1/PD-L2 expression and literature review. Radiat Oncol 13, 167 (2018). https://doi.org/10.1186/s13014-018-1115-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13014-018-1115-x