
Abstract
Background
RAF family activating fusions have been described as a potentially targetable molecular finding in a subset of soft tissue sarcomas. To further expand upon the landscape of this genetic feature, we describe a novel MTAP-RAF1 activating fusion identified in a S100 positive soft tissue sarcoma.
Case presentation
A 51 year old man underwent excision of a soft tissue mass in his foot. Pathology revealed a spindle cell neoplasm with S100 positivity, ultimately classified as a soft tissue sarcoma, not otherwise specified. Comprehensive molecular profiling was performed to help establish the diagnosis and revealed a novel MTAP-RAF1 fusion that includes the tyrosine kinase domain of RAF1.
Conclusions
Our report adds to the spectrum of fusion-driven RAF activation observed in soft tissue sarcomas and lends additional evidence that RAF activation plays an important role in some soft tissue sarcomas. Identification of novel fusions involving the MAPK/ERK pathway in sarcomas may provide new avenues for precision medicine strategies involving targeted kinase inhibitors.
Similar content being viewed by others


Background
Recently, recurrent BRAF gene fusions have been identified in several subgroups of soft tissue sarcomas [1, 2]. To further expand upon the landscape of activating RAF family fusions identified in sarcomas, we report a case of soft tissue sarcoma harboring a novel MTAP-RAF1 fusion. The case was diagnostically challenging as attempts to classify towards a sarcoma subtype were unsuccessful. In addition to sarcoma subtypes, the diagnoses of a spindle cell melanoma variant or clear cell sarcoma were considered given S100 positivity and the biphasic nature of the tumor. In an attempt to aid in the pathological classification of the tumor, comprehensive molecular profiling was performed and interestingly harbored a novel MTAP-RAF1 fusion. To our knowledge, this fusion partnering has not previously been described in the literature to date. Given the potentially high-impact nature and novelty of this finding, we report the clinicopathological details of this case to add to the spectrum of RAF family driven soft tissue sarcomas.
Case presentation
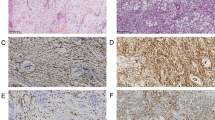
A 51-year-old male presented with a 3 year history of a mass in his right foot with recent enlargement associated with pain. Imaging revealed a soft tissue mass in the plantar arch of the right foot anterior to the calcaneus deep to the fascia. Staging imaging showed no evidence of metastatic disease. The mass was excised. Histologic sections revealed a cellular tumor with scattered ectatic and hyalinized vessels composed of spindle-shaped cells (Fig. 1a) focally arranged in intersecting fascicles admixed with islands of epithelioid cells (Fig. 1b). The spindle cells have vesicular nuclei of variable shape and contour and indistinct cytoplasmic borders with occasional mitoses (Fig. 1c). The epithelioid cells have round to ovoid nuclei with occasional bi-nucleation, and ample eosinophilic cytoplasm, some with focal vacuolation, with distinct cytoplasmic borders (Fig. 1d). No significant necrosis was observed. Immunohistochemical analysis was performed and showed tumor cells positive for S100 (Fig. 1e), specifically the spindle cells were strongly positive while the epithelioid cells were weakly positive. Tumor cells were also positive for TFE3 and vimentin. Immunostains were negative for desmin, MSA, AE1/3, A103, SOX10, MelanA, HMB45, MITF, Tyrosinase and BRAF.

Histological features of the sarcoma. The tumor is composed of (a) spindle-shaped cells focally and (b) arranged in intersecting fascicles with epithelioid islands. c The spindle cells have variable shaped vesicular nuclei with occasional mitoses, (d) whereas the epithelioid cells have round to ovoid nuclei and distinct cytoplasmic borders. e Tumor cells were positive for S100. f Tumor cells show weak nuclear positivity for TFE3
S100 positive, SOX10 negative spindle cell malignancy is a broad pathologic differential. This pattern is observed commonly in a number sarcoma subtypes including synovial sarcoma, Ewing sarcoma, rhabdomyosarcoma, and extraskeletal myxoid chondrosarcoma [3]. S100 positivity is also common in ossifying fibromyxoid tumors, though is typically negative in the malignant cases [4]. SOX10 negative melanoma, clear cell sarcoma, and malignant peripheral nerve sheath tumor (MPNST) are also diagnostic possibilities. We therefore performed additional molecular profiling studies. There was retained nuclear staining of INI-1 and H3K27m3 by immunohistochemistry. Molecular testing performed and interpreted at University of Nebraska, Omaha NE reported the tumor as negative for fusion of the EWSR1 (22q12) and ATF1 (12q13) loci, negative for rearrangement of the TFE3 (Xp11) locus, and negative for fusion of the EWSR1 (22q12) and CREB1 (2q33.3) loci. Additionally, no SS18/SSX1 or SS18/SSX2 fusion transcript was detected by RT-DNA amplification. Given the lack of a more specific molecular finding, the malignant S100-positive tumor was therefore favored to be a soft tissue sarcoma that could not be further subtyped, though could not rule out either a spindle cell melanoma or a fusion-negative clear cell sarcoma.
Given the uncertainty in diagnosis, reference laboratory molecular tumor profiling was performed to identify additional molecular alterations and to potentially aid in patient management. Hybrid capture-based comprehensive next-generation sequencing (Foundation Medicine Inc., Cambridge, MA) revealed a fusion between MTAP exons 1–7 and RAF1 exons 8–17 (Fig. 2). Presence of the fusion was supported by both DNA evidence (150 supporting read pairs) and RNA evidence (437 supporting read pairs). The fusion product disrupts MTAP (S-methyl-5′-thioadenosine phosphorylase) activity but conserves the RAF1 tyrosine kinase domain with loss of the autoinhibitory N-terminal domain [5]. Loss of MTAP, which functions as a tumor suppressor and is vital for methionine salvage, has been observed in a spectrum of cancers including melanoma and sarcoma [6, 7]. Next-generation sequencing (NGS) also reported a low tumor mutation burden of two mutations per megabase and lack of an ultraviolet (UV)-signature as measured by C → T mutations at dipyrimidine sites. Taken in whole, the clinicopathologic and genomic feature of the case including location and size of the mass (deep soft tissue without cutaneous involvement), immunohistochemistry findings, low mutation burden, and lack of a UV-signature ruled against the possibility of this being a melanoma variant. Therefore a fusion-positive soft tissue sarcoma, not otherwise specified, was the final diagnosis.

a Molecular interrogation found a non-reciprocal gene rearrangement involving MTAP on chromosome 9 and RAF1 on chromosome 3. b Supporting RNA sequencing reads spanning the transcriptional breakpoint of the resultant fusion. Sequencing data is visualized in the integrated genomics viewer
Discussion and conclusions
To our knowledge, we report the first observation of an oncogenic MTAP-RAF1 fusion. RAF1 is part of the MAPK/ERK pathway, with the fusion product predicted to upregulate signaling. MTAP has rarely been reported as a partner gene in cancers, typically in relation to intrachromosomal deletion events of the nearby CDKN2A locus [8]. As a known tumor suppressor, loss of MTAP activity due to a fusion event may also contribute to oncogenesis [9]. Recurrent activating BRAF fusions have been reported in several soft tissue sarcoma subtypes [1, 2]. Additionally, BRAF activating mutations have been identified in clear cell sarcomas with observed response to vemurafenib [10, 11]. Our report therefore adds to the spectrum of fusion-driven RAF activation observed in soft tissue sarcomas and lends additional evidence that RAF activation plays an important role in some sarcomas. Though systemic therapy was not indicated in the described case, identification of novel fusions involving the MAPK/ERK pathway in soft tissue sarcomas may provide new avenues for precision medicine strategies involving targeted kinase inhibitors.
Abbreviations
- MTAP:
-
S-methyl-5′-thioadenosine phosphorylase
- NGS:
-
Next generation sequencing
- UV:
-
Ultraviolet
References
Kao YC, Fletcher CDM, Alaggio R, Wexler L, Zhang L, Sung YS, Orhan D, Chang WC, Swanson D, Dickson BC, Antonescu CR. Recurrent BRAF gene fusions in a subset of pediatric spindle cell sarcomas: expanding the genetic Spectrum of tumors with overlapping features with infantile Fibrosarcoma. Am J Surg Pathol. 2018;42:28–38.
Kao YC, Ranucci V, Zhang L, Sung YS, Athanasian EA, Swanson D, Dickson BC, Antonescu CR. Recurrent BRAF gene rearrangements in Myxoinflammatory fibroblastic sarcomas, but not Hemosiderotic Fibrolipomatous tumors. Am J Surg Pathol. 2017;41:1456–65.
Karamchandani JR, Nielsen TO, van de Rijn M, West RB. Sox10 and S100 in the diagnosis of soft-tissue neoplasms. Appl Immunohistochem Mol Morphol. 2012;20:445–50.
Antonescu CR, Sung YS, Chen CL, Zhang L, Chen HW, Singer S, Agaram NP, Sboner A, Fletcher CD. Novel ZC3H7B-BCOR, MEAF6-PHF1, and EPC1-PHF1 fusions in ossifying fibromyxoid tumors--molecular characterization shows genetic overlap with endometrial stromal sarcoma. Genes Chromosomes Cancer. 2014;53:183–93.
Tran NH, Frost JA. Phosphorylation of Raf-1 by p21-activated kinase 1 and Src regulates Raf-1 autoinhibition. J Biol Chem. 2003;278:11221–6.
Li CF, Fang FM, Kung HJ, Chen LT, Wang JW, Tsai JW, Yu SC, Wang YH, Li SH, Huang HY. Downregulated MTAP expression in myxofibrosarcoma: a characterization of inactivating mechanisms, tumor suppressive function, and therapeutic relevance. Oncotarget. 2014;5:11428–41.
Behrmann I, Wallner S, Komyod W, Heinrich PC, Schuierer M, Buettner R, Bosserhoff AK. Characterization of methylthioadenosin phosphorylase (MTAP) expression in malignant melanoma. Am J Pathol. 2003;163:683–90.
Xie H, Rachakonda PS, Heidenreich B, Nagore E, Sucker A, Hemminki K, Schadendorf D, Kumar R. Mapping of deletion breakpoints at the CDKN2A locus in melanoma: detection of MTAP-ANRIL fusion transcripts. Oncotarget. 2016;7:16490–504.
Schmid M, Sen M, Rosenbach MD, Carrera CJ, Friedman H, Carson DA. A methylthioadenosine phosphorylase (MTAP) fusion transcript identifies a new gene on chromosome 9p21 that is frequently deleted in cancer. Oncogene. 2000;19:5747–54.
Park BM, Jin SA, Choi YD, Shin SH, Jung ST, Lee JB, Lee SC, Yun SJ. Two cases of clear cell sarcoma with different clinical and genetic features: cutaneous type with BRAF mutation and subcutaneous type with KIT mutation. Br J Dermatol. 2013;169:1346–52.
Protsenko SA, Semionova AI, Komarov YI, Aleksakhina SN, Ivantsov AO, Iyevleva AG, Imyanitov EN. BRAF-mutated clear cell sarcoma is sensitive to vemurafenib treatment. Investig New Drugs. 2015;33:1136–43.
Acknowledgements
We thank Andrej Savol and Joel Greenbowe for assistance with bioinformatics support for the creation of Fig. 2b.
Availability of data and materials
The datasets generated and analyzed during the current study are not publicly available due patient confidentiality concerns but are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
JKH and ASB conceived and designed the study and were the primary writers of the manuscript. EHJ performed pathological review of the case. JD performed bioinformatics analysis of genomics results. DMJ provided care of the patient and clinical input for the case report. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This retrospective case review was approved by our local institutional review board. The protocol is MCC19161 entitled “Clinical outcomes of the implementation of a personalized medicine clinical service at a major academic cancer center.”
Consent for publication
Written informed consent for publication of their clinical details was obtained from the patient. A copy of the consent form is available for review by the Editor of this journal.
Competing interests
Julia Duggan is an employee of Foundation Medicine, Inc. The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hicks, J.K., Henderson-Jackson, E., Duggan, J. et al. Identification of a novel MTAP-RAF1 fusion in a soft tissue sarcoma. Diagn Pathol 13, 77 (2018). https://doi.org/10.1186/s13000-018-0759-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13000-018-0759-z