
Abstract
A complete genome sequence of human papillomaviruses (HPV) named as HPV-ujs-21015 was determined by viral metagenomic and PCR methods. The complete genome is 7354 bp in length with GC content of 41.7%, of which the genome was predicted to contain six ORFs (Open Reading Frame, ORF) coding for four early proteins (E7, E1, E4, and E2) and two late proteins (L1 and L2). Phylogenetic analysis based on the complete genome and the L1 protein showed that HPV-ujs-21015 belongs to a type 214 member within genus Gamma-6 papillomavirus. It is the first complete genome of Gamma-6 papillomavirus discovered from pregnant women in China.
Similar content being viewed by others


Main text
Human papillomavirus (HPVs), a member of the Papillomaviridae family, are nonenveloped, double-strand circular DNA viruses with an approximately 8 kb genome in length. In the circular genome of HPV, eight genes are typically encoded. L1 and L2 code capsid proteins of virus, which can help virus entry into the basal layer keratinocytes [1, 2]. E2 protein is required for the transcription of viral genes and replication, and also recruits the viral DNA helicase E1 to keeping viral genomes in host cells [3]. E6 and E7 are believed to drive cellular immortalization and maintain the transformed phenotype during tumor progression, to exert functions by binding with many cellular protein to activate cancer hallmarks [4]. HPVs are classified into genera (alpha, beta, gamma, mu, and nu), species, types and even variants based on the nucleotide similarity, with the different types having different life-cycle characteristics and disease associations [5, 6]. HPV persistent infection is the main risk factor for the development of many tumors especially cervical tumor [7]. Although there were numerous ways to prevent the infection of HPV, such as vaccination, over 600,000 cases per year of cervical cancer were recorded worldwide [8]. According to the data from International HPV Reference Center at the Karolinska Institute, Stockholm, Sweden, as of May 6, 2016, two hundred and twenty-six reference HPV types, ranging from HPV-1 to HPV-226, were officially recognized (https://www.hpvcenter.se/human_reference_clones/). The determination of HPV genome can be helpful to understand the genomic characteristics and the clinical relevance of these new HPV strains. In recent years, in addition to frequently-used methods like PCR, some new methods including viral metagenomics were used to acquire the genome of HPV more efficiently [9, 10].
In our current study, the viral nucleic acid sequences from vaginal swabs were investigated through viral metagenomics. A total of 100 vaginal swabs were collected from the health pregnant women who visited hospital for antenatal follow-up of pregnancy in Shanghai City, China, in 2017. The total viral nucleic acid was isolated using QiaAmp Mini Viral RNA kit (Qiagen, USA) according to the manufacturer’s protocol after centrifugation, filtration and DNase and RNase digestion, as we described previously, and pooled into 10 libraries [9]. The produced nucleic acids (both DNA and RNA) were subjected to reverse transcript with N8 random primers (Sangon, Shanghai, China), and the second stand was generated using Klenow enzyme (NEB, Ipswich, USA). The libraries were then constructed by the Nextera XT DNA sample Preparation Kit (Illumina, CA, USA) following the protocol, and the prepared libraries were sequenced by Illumina Miseq platform with 250 bases paired ends with dual barcoding for each pool.
The total numbers of sequence reads generated for the 10 libraries were 73,264 (swab01), 45,462 (swab02), 100,518 (swab03), 111,398 (swab04), 82,612 (swab05), 436,560 (swab06), 903,618 (swab07), 71,544 (swab08), 273,046 (swab09), and 51,590 (swab10). Raw data were processed according to the standard procedure which included debarcoding, trimming and assembling [11]. Contigs and singlet reads were then matched against a customized viral proteome database using BLASTx with an E value cutoff of < 10− 5. Bioinformatics analysis was performed according to a previous study [9]. PCR and sanger sequencing were carried out to bridge the gaps between sequences as well as assess the prevalence of HPV strain identified in this study. Putative ORFs (Open Reading Frame, ORF) in the genome of HPV-ujs-21015 were predicted by Geneious Prime software (version 2020.0.4). The closest viral strains based on best BLASTx hits and the representative members of species and genera were selected to perform the phylogenetic analyses (Table 1). In order to construct the phylogenetic tree, sequence alignment was performed using Clustal W with the default settings. Phylogenetic tree was generated using the maximum likelihood method based on Jones-Taylor-Thornton (JTT) model by MEGA 7.0 with 1000 bootstrap. Bootstrap values for each node are given in the trees.
Results and discussion
A strain of HPV named as HPV-ujs-21015 (GenBank accession no. MN400665, see Additional file 1) was determined in the vaginal swab (containing 1654 reads in library swab02), of which the complete genome is 7354 bp in length with GC content of 41.7%. The genome of HPV-ujs-21015 was predicted to contain six ORFs coding for four early proteins (E7, E1, E4, and E2) and two late proteins (L1 and L2) (Fig. 1). The nucleic acid lengths of these proteins were 300, 1905, 354, 1167, 1626 and 1614, respectively, and the positions on the genome were showed in Fig. 1. Notably, the E6 gene that plays a crucial role in the cell transformation through binding of p53 tumor suppressor protein was absent in this strain, which was consistence with other HPV214 strains [12, 13]. E6 as well as E7 is believed to be directly responsible for the development of HPV-induced carcinogenesis. In the high risk HPVs, they do this cooperatively by targeting diverse cellular pathways including the regulation of cell cycle control. Meanwhile, there is a view that the lost function of E6 in HPV214 may be compensated for in its E7 protein which has an LXCXE (Fig. 2a) motif that has been shown to bind pRB in HPV16 and other high risk HPV types.

Genomic organization of HPV-ujs-21015. The genomic positions of viral genes (E7, E1, E2, E4, L1 and L2) were indicated in the figure

Alignment of amino acid of E7 (a) and L1 proteins (b) between HPV-UJS-2015 and Related HPVs. The LXCXE and Zinc-finger domains were enclosed with solid or dotted box, respectively. Mutation and deletion were marked with solid or blank stars
According to the International Committee on Taxonomy of Viruses (ICTV), a viral type within a species has 71 to 89% identity with other types within the same species based on the comparative homology of the L1 DNA sequence. Additionally, there are several subtypes and variants within a type, which share 90 to 98% and more than 98% identity, respectively. In the current study, sequence analysis indicated that HPV-ujs-21015 shared the highest nucleotide (nt) sequence identity (99%) with a type 214 strain named CT06 isolated from South African strain (GenBank no. MF509819), as well as strain mw03c65 (GenBank accession no. MF588697), which was an unclassified strain detected in patients with immunodeficiency in USA.
Similar to mw03c65 and CT06 strain, the putative E7 protein of HPV-ujs-21015 strain contained one zinc-finger domain and an LXCXE sequence (Fig. 2a), which is critical for transforming activities by way of binding a number of important cellular regulatory proteins, including tumor suppressor: Retinoblastoma protein (pRb). Compared with these two strains, HPV-ujs-21015 had one amino acid deletion and three mutations (Fig. 2a). Whether the deletion and mutations affect the biological function of E7 will require more research. Intriguingly, another protein with significant diversity was L1, of which HPV-ujs-21015 had the 100% amino acid similarity with mw03c65, but was thirty consensus amino acid longer than that of CT06 strain in the 5’end (Fig. 2b).
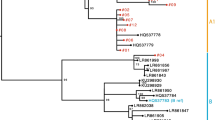
To characterize the phylogenetic relationship between HPV-ujs-21015 and related HPV reference strains, two phylogenetic trees based on the complete genome and L1 protein were constructed, respectively, by MEGA 7.0. Both trees revealed that the reference HPVs were clustered well in their genera and types. The phylogenetic tree based on the complete genome showed that HPV-ujs-21015 belonged to Gamma-papillomavirus (Fig. 3a). The other phylogenetic tree based on the L1 protein further assigned HPV-ujs-21015 within the group of type 214 in Gamma-6, being closely related to mw03c65 strain (Fig. 3b). In summary, our results suggest that all of these three strains isolated from different countries were variants with the genotype 214.

Phylogenetic trees constructed based on the complete genome (a) and L1 protein (b) were constructed, respectively, using maximum-likelihood method by MEGA-X with 1000 bootstrap. GenBank accession nos. of the reference strains and their abbrevation were showed in the trees. The strain determined in this study was marked with a triangle
HPVs comprise five evolutionary groups with different epithelial tropisms and disease associations. Traditionally, based on the location of the certain virus genome was found, HPVs have also been classified as mucosal or cutaneous types [1]. Increasing evidences revealed that Gamma-PVs showed broad tissue tropism, with the detection locations ranging from health skin and cutaneous lesions to genital lesions [10, 14, 15]. DNA of some Gamma-PVs types were detected in skin cancer raised concerns of some Gamma-PVs associations with cancers, especially in patients with immunodeficiency or immunosuppression [16, 17]. In the current study, HPV-ujs-21015 strain was identified from a health pregnant woman who visited hospital for antenatal follow-up. Vaginitis or other vaginal disease were not found by the attending gynecologist. Generally, both mucosal or cutaneous disease relied on the persistent infection of HPVs. Therefore, whether the infection of HPV-ujs-21015 can cause disease or not is still unknown. A total of one hundred of vaginal swab samples from health pregnant women who visited hospital for antenatal follow-up were screened by PCR method with a set of nested primers (data not showed) designed on HPV-ujs-21015 L1 gene. Result showed that two samples were positive (2/100). The prevalence and disease association of HPV-ujs-21015 need to be clarified through larger sample size, biological and histological experiments.
In conclusion, we determined and characterized the complete genome sequence of a genotype 214 Gamma-6 papillomavirus, which was isolated from a health pregnant woman of China. To the best of our knowledge, it is the first complete genome of Gamma-6 papillomavirus detected in Pregnant Women of China.
Availability of data and materials
The sequences of full-length envelope gene generated in this study have been deposited in GenBank under the accession numbers MN400665.
Abbreviations
- aa:
-
Amino acid
- HPV:
-
Human papillomaviruses
- ORF:
-
Open Reading Frame
- ICTV:
-
International Committee on Taxonomy of Viruses
- nt:
-
Nucleotide
- pRb:
-
Retinoblastoma protein
References
Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, Stanley MA. The biology and life-cycle of human papillomaviruses. Vaccine. 2012;30(Suppl 5):F55–70.
Finnen RL, Erickson KD, Chen XJS, Garcea RL. Interactions between papillomavirus L1 and L2 capsid proteins. J Virol. 2003;77(8):4818–26.
Thomas Y, Androphy EJ. Acetylation of E2 by P300 Mediates Topoisomerase Entry at the Papillomavirus Replicon. J Virol. 2019;93(7):e02224–18.
Hoppe-Seyler K, Bossler F, Braun JA, Herrmann AL, Hoppe-Seyler F. The HPV E6/E7 oncogenes: key factors for viral carcinogenesis and therapeutic targets. Trends Microbiol. 2018;26(2):158–68.
Bernard HU, Burk RD, Chen ZG, van Doorslaer K, zur Hausen H, de Villiers EM. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology. 2010;401(1):70–9.
Bosch FX, Burchell AN, Schiffman M, Giuliano AR, de Sanjose S, Bruni L, Tortolero-Luna G, Kjaer SK, Munoz N. Epidemiology and natural history of human papillomavirus infections and type-specific implications in cervical Neoplasia. Vaccine. 2008;26:K1–K16.
Schwarz TF. AS04-adjuvanted human papillomavirus-16/18 vaccination: recent advances in cervical cancer prevention. Expert Rev Vaccines. 2008;7(10):1465–73.
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–917.
Liu ZJ, Yang SX, Wang Y, Shen Q, Yang Y, Deng XT, Zhang W, Delwart E. Identification of a novel human papillomavirus by metagenomic analysis of vaginal swab samples from pregnant women. Virol J. 2016;13:122.
Pastrana DV, Peretti A, Welch NL, Borgogna C, Olivero C, Badolato R, Notarangelo LD, Gariglio M, FitzGerald PC, McIntosh CE, et al. Metagenomic Discovery of 83 New Human Papillomavirus Types in Patients with Immunodeficiency. Msphere. 2018;3(6).
Deng X, Naccache SN, Ng T, Federman S, Li L, Chiu CY, Delwart EL. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res. 2015;43(7):e46.
Murahwa AT, Meiring TL, Mbulawa ZZA, Williamson AL. Discovery, characterisation and genomic variation of six novel Gammapapillomavirus types from penile swabs in South Africa. Papillomavirus Res. 2019;7:102–11.
Nobre RJ, Herraez-Hernandez E, Fei JW, Langbein L, Kaden S, Grone HJ, de Villiers EM. E7 oncoprotein of novel human papillomavirus type 108 lacking the E6 gene induces dysplasia in organotypic keratinocyte cultures. J Virol. 2009;83(7):2907–16.
Bolatti EM, Hosnjak L, Chouhy D, Re-Louhau MF, Casal PE, Bottai H, Kocjan BJ, Stella EJ, Gorosito MD, Sanchez A, et al. High prevalence of Gammapapillomaviruses (gamma-PVs) in pre-malignant cutaneous lesions of immunocompetent individuals using a new broad-spectrum primer system, and identification of HPV210, a novel gamma-PV type. Virology. 2018;525:182–91.
Bolatti EM, Chouhy D, Casal PE, Perez GR, Stella EJ, Sanchez A, Gorosito M, Bussy RF, Giri AA. Characterization of novel human papillomavirus types 157, 158 and 205 from healthy skin and recombination analysis in genus gamma-papillomavirus. Infect Genet Evol. 2016;42:20–9.
Bottalico D, Chen ZG, Dunne A, Ostoloza J, McKinney S, Sun C, Schlecht NF, Fatahzadeh M, Herrero R, Schiffman M, et al. The Oral cavity contains abundant known and novel human papillomaviruses from the Betapapillomavirus and Gammapapillomavirus genera. J Infect Dis. 2011;204(5):787–92.
Dutta S, Robitaille A, Aubin F, Fouere S, Galicier L, Boutboul D, Luzi F, Di Bonito P, Tommasino M, Gheit T. Identification and characterization of two novel Gammapapillomavirus genomes in skin of an immunosuppressed Epidermodysplasia Verruciformis patient. Virus Res. 2018;249:66–8.
Acknowledgements
Not applicable.
Funding
This study was financially supported by Changzhou Social Development Project (Grant No. CE20185002); Jiangsu social development project (No. BE2016663), Zhangjiagang science and technology planned project (ZKS1703), Research Funds of Jiangsu Entry-Exit Inspection and Quarantine Bureau (No. 2018KJ07).
Author information
Authors and Affiliations
Contributions
QS and WZ conceived the study and designed the experiments. YL, JQW, JY, JPX and YFW collected the samples, YL and RZ and JL performed the laboratory assays. SXY and XCW contributed to analysis of sequencing data. LY wrote the initial draft, and QS edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study did not include experiments with human participants or animals performed by any of the authors.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
The complete genome of HPV-ujs-21015.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ling, Y., Wang, J., Yin, J. et al. Genomic organization of a Gamma-6 papillomavirus metagenomic discovered from vaginal swab samples of Chinese pregnant women. Virol J 17, 44 (2020). https://doi.org/10.1186/s12985-020-01319-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-020-01319-9