
Abstract
Background
The picornaviral 3C protease mediates viral polyprotein maturation and multiple cleavages of host proteins to modulate viral translation and transcription. The 3C protease has been regarded as a valid target due to its structural similarity among different picornaviruses and minimal sequence similarity with host proteins; therefore, the development of potent inhibitors against the 3C protease as an antiviral drug is ongoing. Duck hepatitis A virus (DHAV) belongs to the Picornavidea family and is a major threat to the poultry industry. To date, little is known about the roles of the DHAV 3C protease plays during infection.
Methods
In this study, we compared the full-length DHAV 3C protein sequence with other 3C sequences to obtain an alignment for the construction of a phylogenetic tree. Then, we expressed and purified recombinant DHAV 3C protease in the BL21 expression system using nickel-NTA affinity chromatography. The optimization of the cleavage assay conditions and the kinetic analysis for DHAV 3C protease were done by in vitro cleavage assays with a fluorogenic peptide respectively. The inhibitory activity of rupintrivir against the DHAV 3C protease was further evaluated. The localization of the 3C protease in infected and transfected cells was determined using immunofluorescence and confocal microscopy.
Results
Under different expression conditions, the 3C protease was found to be highly expressed after induction with 1 mM IPTG at 16 °C for 10 h. We synthesized a fluorogenic peptide derived from the cleavage site of the DHAV polyprotein and evaluated the protease activity of the DHAV 3C protease for the first time. We used fluorimetric based kinetic analysis to determine kinetic parameters, and Vmax and Km values were determined to be 16.52 nmol/min and 50.78 μM, respectively. Rupintrivir was found to exhibit inhibitory activity against the DHAV 3C protease. Using polyclonal antibody and an indirect immunofluorescence microscopy assay (IFA), it was determined that the DHAV 3C protease was found in the nucleus during infection. In addition, the DHAV 3C protease can enter into the nucleus without the cooperation of viral proteins.
Conclusions
This is the first study to examine the activity of the DHAV 3C protease, and the activity of the DHAV 3C protease is temperature-, pH- and NaCl concentration- dependent. The DHAV 3C protease localizes throughout DHAV-infected cells and can enter into the nucleus in the absence of other viral proteins. The kinetic analysis was calculated, and the Vmax and Km values were 16.52 nmol/min and 50.78 μM, respectively, using the Lineweaver–Burk plot.
Similar content being viewed by others

Background
Duck hepatitis A virus (DHAV) causes an acute, contagious, and highly fatal disease that is characterized by a swollen liver mottled with hemorrhages and effects young (less than 3 weeks old) ducklings [1]. In contrast, adult ducks infected with DHAV do not become clinically ill, however, infection induces the egg drop syndrome [2]. DHAV was first described in the United States and isolated from chick embryos in 1949 [3]. Subsequently, outbreaks of duck viral hepatitis in other parts of the world including China, South Korea, and Japan were reported [4,5,6]. Birds are a well-known reservoir of infectious diseases. Due to the high mortality associated with the disease, DHAV causes significant financial losses, while no public health concern has been identified [3]. A previous study showed that natural infection occurs in domestic ducks [3]. DHAV infection was recently identified in mallards, goslings and pigeons [7]. A goose embryonated epithelial cell line efficiently assists with the replication of DHAV, which shows a higher virus titer compared to other duck cell lines [8]. Based on phylogenetic and neutralization assays, DHAV has been divided into three distinct genotypes: DHAV-1, DHAV-2, and DHAV-3 [1, 9]. DHAV-1 is the most common type and has spread worldwide, and the development of accurate detection methods is essential [10,11,12,13,14].
According to the International Committee on Taxonomy of Viruses (ICTV) report, DHAV belongs to the genus Avihepatovirus, as a member of the family Picornaviridae [15]. DHAV is a small, simple, nonenveloped, spherical icosahedral virus that is approximately 30 nm in diameter and contains a single-stranded positive-sense RNA genome of approximately 7.7 kb. The viral genome contains one open reading frame (ORF) that encodes a single polyprotein including structural proteins, P1 region (VP4/VP2/VP3/VP1), and nonstructural proteins, P2 (2A1/2A2/2A3/2B/2C) and P3 (3A/3B/3C/3D) regions, as well as two untranslated regions (5′ UTR and 3′ UTR) [16]. The DHAV 3D protein was confirmed to recognize and bind the 3′ UTR as an RNA-dependent RNA polymerase (RdRp) [17]. The processing of the polyprotein depends on viral proteases to produce functional and mature proteins. In general, the leader protease in aphthovirus, 2A protease in enteroviruses, and 3C protease in most picornaviruses contribute to the processing of the polyprotein [18, 19]. In contrast to the highly nonconserved 2A proteins in the family Picornaviridae, the 3C protease is a conserved chymotrypsin-like serine protease with a conserved catalytic triad of His-Asp-Cys. Furthermore, due to its structural conservation, the 3C protease is an important antiviral target for the development of inhibitors [20,21,22,23]. Regarding DHAV, it does not possess an L protein but has three 2A proteins. Specifically, DHAV possesses an NPG/P autocleavage motif in 2A1, avrRpt2-induced gene 1 (AIG1) domain in 2A2, and H-NC motif in 2A3 protein [24, 25]. It is likely that the 3C protease is an important putative protease in DHAV for polyprotein processing [26] (Table 1).
In other reported viruses, the 3C protease targets certain cellular factors for efficient viral infection. For instance, the 3C protease could mediate the cleavage of cellular proteins such as cAMP response element-binding protein-1(CREB-1), polyadenylation factor (CstF-64), and TATA box binding protein (TBP), which are associated with the blockage of host genome transcription [27,28,29]. Furthermore, the 3C protease is capable of disrupting intrinsic immune responses by inhibiting the functions of some immune factors. During enterovirus infection, the 3C protease induces cleavage of TIR-domain-containing adapter-inducing interferon-β (TRIF), interferon regulatory factor 7 (IRF7) and transforming growth factor-β-activated kinase 1 (TAK1) to escape host antiviral signaling [30,31,32]. In coxsackievirus B3 (CVB3) infection, the 3C protease cleaves TRIF and mitochondrial antiviral signaling protein (MAVS) to suppress host immunity [33]. In DHAV infection, it has been reported that activation of the toll-like receptor 7 (TLR7) pathway is involved in the immune response and viral clearance [34]. To date, some epidemiological, pathogenic and immunological mechanisms of DHAV have been reported [35,36,37]. It is likely that different picornaviruses adopt various strategies to interfere with cellular resources.
Fluorescence resonance energy transfer (FRET)-based assays have been utilized successfully to monitor enzyme activity using substrates composed of peptides that are known to be recognized and cleaved by a protease. To date, the kinetics of some picornaviral proteases have been characterized, including human rhinovirus (HRV), enterovirus 71 (EV71), hepatitis A virus (HAV) and foot-and-mouth disease virus (FMDV), providing insight into the proteolytic mechanism and inhibitor discovery. The EV71 3C protease exhibited the protease activity (Km = 30 ± 2 μM, Vmax = 85 ± 1.4 nM min− 1) against the 3B-3C cleavage site [38]. In addition, the enzyme activity of the EV71 3C protease was increased to 60-fold (Km = 5.8 ± 0.9 μM) against a substrate of the SARV-CoV 3C-like protease compared to the autoprocessing site (VP2-VP3, VP3-VP1, 2A-3C, 3A-3B, 3B-3C, and 3C-3D) [39]. For FMDV, different mutations in the cysteine residue at position 142 resulted in different reductions in the protease activity of the 3C protease compared to wild type (kcat/Km = 990 ± 20 M− 1 s− 1) [40]. However, studies on the DHAV 3C protease have been limited [41]. Here, we report the activity and localization of the DHAV 3C protease, which provides insight for a better understanding and further characterization of the 3C protease in DHAV infection.
Methods
Cells and virus
The separation and preservation of the DHAV H strain were performed at the Institute of Preventive Veterinary Medicine of Sichuan Agricultural University. Duck embryo fibroblast (DEF) cells were cultured in modified Eagle’s medium (MEM; Gibco) containing 10% newborn bovine serum (NBS; Gibco) and cultured in a humidified 37 °C, 5% CO2 incubator.
Construction of the pEGFP-3C recombinant and mutant plasmids
The pEGFP N1 and pMD19-T simple/DHAV 3C plasmids were digested with EcoR I and BamH I (Takara) at 37 °C to generate fragments. The DNA sequence encoding DHAV 3C (181 aa, Table 1) was fused with the green fluorescent protein (GFP) sequence at the N-terminal through ligation. The newly synthesized pEGFP DHAV 3C plasmid was then used for site-directed mutagenesis to alter the catalytic triads of 3C such that the histidine at position 38 or the cysteine at position 144 was substituted with an alanine. The 3C sequence was cloned into the pcDNA 3.1/myc-His (−) vector for expression. All constructs were verified by DNA sequencing. The resulting plasmids, pEGFP-3C, pEGFP-3C-H38A, and pEGFP-3C-C144A, were used for the expression of the fusion proteins.
Evolutionary analysis of the picornaviral 3C protease
The protein sequences of the 3C protease were searched from GenBank in the National Center for Biotechnology Information (NCBI) database. There were eighty protein sequences of different single-stranded RNA viruses, including picornaviruses and dicistroviruses. The sequence alignment was performed by ClustalW in MEGA 7.0 software. The phylogenetic relationship between these protein sequences was analyzed by the maximum likelihood method using MEGA 7.0 software with 1000 bootstrap replicates and visualized with iTOL.
Transfection of plasmid DNA
DEF cells grew to 70–80% confluence in MEM at 37 °C before transfection with plasmid DNA. According to the manufacturer’s standard protocol, transfection was performed with Lipofectamine 2000 reagent (Invitrogen). After 24 h of transfection, the cells were washed with phosphate-buffered saline (PBS) three times and treated with 4′6-diamidino-2- phenylindole (DAPI; Beyotime).
Expression and purification of the DHAV 3C protease
The amplification of the 3C gene was performed after using DHAV RNA (DHAV-H; YP_007969882.1) as a template to perform reverse transcription. It was cloned into the Nde I and Hind III sites of the pET32a vector to construct the DHAV 3C protease linked with a hexahistidine tag at its N-terminus (primer sequences are shown in Table 2). The recombinant plasmid was sequenced for verification to ensure no mutations. Subsequently, Escherichia coli BL21 cells were transformed with plasmids for expression. The positive BL21 cells were cultured in Luria-Bertani (LB) medium containing 100 mg/liter ampicillin at 37 °C. Protein expression was induced with isopropyl-β-D-thiogalactopyranoside (IPTG) at different concentrations for 12 h at 16 °C when the optical density at 600 nm (OD600) of the culture reached 0.6. Cells were harvested and then resuspended in lysis buffer [50 mM NaH2PO4·2H2O (pH 8.0) and 300 mM NaCl]. Subsequently, the cells were ultrasonically lysed on ice. After centrifugation at 10,000×g for 20 min at 4 °C, the supernatant of the lysate was loaded into an equilibrated nickel ion-agarose affinity chromatography. Ni-NTA was washed with lysis buffer containing imidazole at low concentrations (10 mM and 20 mM), and nonspecific proteins were removed by washing. Subsequently, the 3C protease was eluted with lysis buffer containing 100 mM imidazole. Then the purified 3C protease was concentrated at 4 °C to 2 mg/ml in a reaction buffer [150 mM NaCl, 20 mM Tris-HCl (pH 7.0), 5 mM DTT (pH 5.2), 1 mM EDTA, and 10% glycerol]. All 3C protein was stored at − 80 °C until use. Statistical analysis was performed with GraphPad Prism 5 software.
Western blot analysis
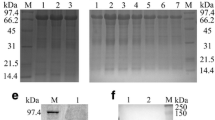
Recombinant proteins were separated by SDS-PAGE and transferred onto a polyvinylidene fluoride (PVDF) membrane. Subsequently, the PVDF membranes were blocked with 5% skim milk at 37 °C for 1 h and then incubated with rabbit anti-DHAV serum (1:500 dilution) at 4 °C overnight. Then, the membranes were washed with TBST (containing 0.1% Tween-20) three times and incubated with goat anti-rabbit antibody (Bio-Rad, 1:5000) at 37 °C for 1 h respectively. After that, the membranes were washed in PBST and treated with Western BLoT Chemiluminescence HRP Substrate (TaKaRa) for the detection of specific bands. The bacterial lysate of the empty vector was used as a control group.
Preparation of polyclonal antibody
The purified DHAV 3C protease was used to generate a polyclonal antibody. Approximately 20 μg 3C protein and an equal volume of Quick Antibody adjuvant (Biodragon-Immunotech) were mixed and then injected into 6-week-old female BALB/c mice. After 2 weeks, a booster immunization was needed in the same manner. Using an indirect enzyme-linked immunosorbent assay (ELISA), serum antibody titers were measured, and then anti-serum was collected from the eyeballs of mice and frozen at − 80 °C.
Indirect immunofluorescence microscopy (IFA)
DEF cells were grown to 50–70% confluence on glass coverslips as a monolayer before being infected with DHAV (1000 TCID50). The cells were fixed at appropriate intervals postinfection in 4% paraformaldehyde for 60 min and then rinsed with PBS. DEF cells were permeabilized with 0.2% Triton X-100 for 30 min at room temperature and incubated with 5% BSA blocking solution for 60 min followed by overnight incubation with anti-3C antibody. Then, the cells were incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG for 60 min. After that, the cells were washed in PBST and treated with DAPI (Beyotime). A confocal microscope (Nikon A1) was used to capture images.
Synthesis of substrate peptides
The synthetic peptide substrate that was tested with a high-performance liquid chromatography (HPLC)-based assay was purchased from TOP Biochem (Shanghai, China). The synthetic peptide substrates were attached with a fluorescence quenching pair, 5′-[(2-aminoethyl) amino] naphthalene-1-sulfonic acid (Edans) and p-(p-dimethylaminophenylazo) benzoic acid (Dabcyl) as a donor and a quencher moiety, respectively. Corresponding to the DHAV-H polyprotein processing site between 2C–3A, the fluorogenic peptide Dabcyl-ASFMNQSKVRRFE-Edans (96% purity) was designed.
In vitro cleavage assay
Fluorescence experiments were performed with Varioskan® Flash. The determination of the cleavage activity of the DHAV 3C protease was performed in 100 μl reaction buffer [150 mM NaCl, 20 mM Tris-HCl (pH 7.0), 5 mM DTT (pH 5.2), 1 mM EDTA, and 10% glycerol] containing 50 μl enzyme (10 μM) and 50 μl peptides (2 μM to 300 μM). Reaction mixtures were incubated in a black 96-well microplate at 37 °C. When the protease cleaved the quencher bond, the fluorophore separated from the fluorescence quencher moiety enabling fluorescence to be detected. The relative fluorescence reflected the degree of protease activity. The relationship between substrate concentration and the relative fluorescent unit (RFU) was measured with the Edans standard. The RFU measurements were collected using an excitation wavelength of 340 nm and an emission wavelength of 490 nm. Lineweaver-Burk’s method was applied to calculate Km and Vmax. The protease and substrate were incubated respectively at five different temperatures (4, 16, 30, 37, and 50 °C). In addition, the reaction sample was carried out at different pH values (5, 6, 7, 8, 9, and 10) or with different NaCl concentrations in buffer (50, 100, 150, 200, and 500 mM) to examine the optimal cleavage reaction conditions for the DHAV 3C protease. The fluorescence of the reaction was detected following incubation at 37 °C for 2 h. Triplicate reactions were carried out to identify the effects of temperature, NaCl concentration, and pH value on the activity of the DHAV 3C protease.
Results
Evolutionary analysis
Based on the amino acid sequence alignment of the 3C proteases of different picornaviruses, the catalytic triad of His-Glu/Asp-Cys was highly conserved among the 3C proteases of all selected viruses (Fig. 1a). Phylogenetic analysis revealed that the 3C proteases from picornaviruses were divided into six major groups (Fig. 1b). The 3C protease of the Avihepatovirus family was located at the same branch with four other avian picornaviruses (duck aalivirus, chicken picornavirus 2, turkey avisivirus and chicken picornavirus 3) and are highlighted in red. The 3C gene within these viruses shared 38.32–45.52% homology at the amino acid level, which has higher sequence similarities comparing with other picornaviruses. This result is different from the phylogenetic trees of the P1 region and 3D polymerase, which showed scattered pattern of avian picornaviruses [42].

Multiple sequence alignment of 3C proteases from various picornaviruses. a Alignment of DHAV and several other picornavirus amino acid sequences corresponding to the 3C protease. Invariant residues in the 3C protease are highlighted with a black background. The catalytic triad of the 3C protease is indicated with black arrows. The GxCG motif was conserved within these sequences, which may be part of the active site. b Phylogenic tree of the 3C proteases. The tree contains 80 sequences that correspond to 3C protein sequences from different single-stranded RNA viruses. Phylogenetic analysis was predicted with the maximum likelihood method. The background colors indicate that these 3C proteases were divided into six groups. The stability of the nodes was assessed with 1000 bootstrap replications, and bootstrap values above 0.5 were shown at the blue circles. The width of the blue circles represents the bootstrap value (range from 0.5 to 1)
Expression and purification of the DHAV 3C protease
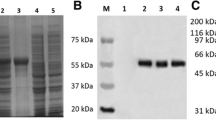
The 3C gene was cloned into the Nde I-Hind III-digested vector pET32a and then expressed under different conditions, including induction temperatures, inducer concentrations, and induction durations (Fig. 2). The expression products were fractionated with SDS-PAGE and compared with E. coli cells containing a pET32a vector. An additional distinct band was observed with a molecular mass of 20.6 kDa when pET32a-3C expression was induced with IPTG at 16 °C (Fig. 2b). In addition, the expression condition of the DHAV 3C protease was optimized when induced using 1 mM IPTG at 16 °C for 10 h (Fig. 2a and c). The recombinant 3C protease was expressed with a hexahistidine tag at the N-terminal and therefore purified with Ni2+-NTA. The purified 3C protease was used to produce an anti-3C polyclonal antibody. The results of Western blotting indicated that the anti-DHAV duck serum could react with the recombinant 3C protease, showing a specific band corresponding to approximately 20 kDa (Fig. 2d).

Optimization of 3C protease expression. The DHAV 3C protease was expressed at different (a) IPTG concentrations (0.2, 0.4, 0.6, 0.8, 1.0, 1.2, and 1.4 mM), (b) temperatures (16, 30, and 37 °C), and (c) induction times (2, 4, 6, 8, 10, and 12 h). (d) The bacterial lysates were analyzed for the presence of the DHAV 3C protease by immunoblotting
Optimization of cleavage assay conditions
The fluorometric substrate was cleaved by the 3C protease at the 2C-3A junction, which was derived from the native protease cleavage site within the DHAV polyprotein. The effect of pH on the enzymatic activity of the DHAV 3C protease was determined when reaction buffers in the pH range from pH 5 to 10 were used (Fig. 3a). The results showed that the optimal pH for the DHAV 3C protease was pH 7.0, similar to previous reports on the enteroviral 3C protease. Notably, the enzymatic activity of the 3C protease was reduced when the pH was decreased to pH 5. At optimal pH, the maximum fluorescence was observed at a concentration of 150 mM NaCl (Fig. 3b). The optimal temperature for enzymatic activity and substrate stability was observed with incubation at 37 °C. The results from different reactions showed that the protease activity decreased at temperatures below 37 °C, while substrate stability decreased at 50 °C (Fig. 3c). Consequently, the optimal temperature for 3C protease activity was regarded as 37 °C. These cleavage reaction conditions were utilized in downstream assays.

Analysis of the DHAV 3C protease activities under different conditions. Different (a) pH values of the reaction buffer (b) NaCl concentrations, and (c) temperatures
Kinetic analysis of DHAV 3C protease substrate
To measure the substrate specificity and cleavage efficiency of the DHAV 3C protease in vitro, the kinetic parameters for cleavage of this peptide substrate (Dabcyl-ASFMNQSKVRRFE-Edans) were determined. The Vmax and Km values of the DHAV 3C protease were determined by measuring the activity with various substrate concentrations ranging from 10 μM to 300 μM (Fig. 4). The kinetic analysis was calculated using the Lineweaver–Burk plot. The Vmax and Km values were 16.52 nmol/min and 50.78 μM, respectively.

In vitro cleavage activity of the DHAV 3C protease. The cleavage reactions were performed with different concentrations of substrate. The enzymatic activity of the DHAV 3C protease was determined by incubation with (a) 10 μM, (b) 100 μM, (c) 150 μM, (d) 200 μM, (e) 250 μM, and (f) 300 μM substrate. All reactions were determined in triplicate and kinetic parameters were calculated using GraphPad Prism 5
Z-factor analysis
The Z-factor value was required to evaluate the quality of the fluorogenic substrate-based assay, which was used to screen inhibitors of the DHAV 3C protease. A Z-factor value in a range of 0.5 to 1.0 indicates that the assay is suitable for high-throughput screening assays [43]. The Z-factor was determined from 60 independent samples as positive controls containing 5 μM substrate and 5 μM 3C protease and 60 independent samples as negative controls containing 5 μM substrate without protease (Table 3). The assay range and data derivation are shown in a scatter plot using the relative fluorescence of the positive control and negative control (Fig. 5a). The Z-factor was calculated as 0.548336, which indicated that the quality of the assay was reliable, and further high-throughput screening assays were required.

The inhibition of AG7088 against the DHAV 3C protease. (a) The Z-factor assays of the DHAV 3C protease. The Z-factor assays were performed with positive groups (n = 60) containing 5 μM substrate and 5 μM DHAV 3C protease and negative groups (n = 60) consisting of 5 μM substrate and reaction buffer. (b) The inhibition assay was performed with samples containing 5 μM DHAV 3C protease, 20 μM substrate and two concentrations (50 μM and 100 μM) of the AG7088 in cleavage buffer
Evaluation of the inhibitor
Based on the established FRET substrate-based assay for the DHAV 3C protease, the HRV 3C protease inhibitor Rupintrivir (AG7088) was purchased to detect its inhibitory activity against the DHAV 3C protease. As described above, the protease activity of the DHAV 3C protease was monitored during incubation with different concentrations of the inhibitor. The results showed that AG7088 exhibited inhibitory activity against the DHAV 3C protease (Fig. 5b).
Localization of the 3C protease
The 3C protease in some picornaviruses has been shown to cleave host transcription and translational factors in the nuclei. Figure 6 shows the localization of the 3C protease in DEF cells infected with DHAV at 3, 6, and 9 h postinfection (hpi). This 3C antibody could not react with mock-infected cells, and the signal in infected cells colocalized with a FITC-UTP stain that was added to mark the localization of the 3C protease. The 3C protease was observed in the cytoplasm as early as 3 hpi. At 6 hpi, strong fluorescence corresponding to the 3C protease was detected in the nucleus. It seemed that the DHAV 3C protease had the ability to potentially target the nucleus. To confirm whether the 3C protease entered into the nucleus directly or through a 3C-containing precursor, we cloned the 3C sequence and 3C variants into a pEGFP-N1 vector. The nucleotide sequences of the 3C variants encoding for H38 and C144 were mutated into sequences encoding alanine residues to create single variants of the 3C protease. Then, the N-terminally GFP-tagged 3C protease and variants were expressed as in DEF cells. As shown in Fig. 8A, it was observed that the fluorescence of EGFP-3C and its variants were distributed throughout the cell cytoplasm and nucleus. An indirect immunofluorescence microscopy (IFA) assay was carried out to verify that the fluorescence came from the 3C fusion proteins instead of free GFP. The results showed that the 3C fusion proteins were correctly expressed in DEF cells (Fig. 7a). Notably, some fluorescent signals were generated in the cytoplasm of cells expressing EGFP-3C-H38A and EGFP-3C-C144A. Untagged 3C was located in the cytoplasm and nucleus, indicating that the ability to enter into the nucleus was not the result of GFP expression (Fig. 7b). Taken together, these results indicated that the DHAV 3C protease had intrinsic nuclear-targeting potential.

Localization of the 3C protease in DEF cells infected with DHAV. DEF cells were infected with DHAV and collected at various times postinfection. The samples from each time point were fixed with 4% paraformaldehyde and stained with DAPI for DNA (blue) and 3C antibody (green) as indicated

The 3C protease and variants were expressed in DEF cells. a DEF cells were transfected with pEGFP-3C, pEGFP-3C-H38A, pEGFP-3C-C144A, and pEGFP-N1 for 24 h. DEF cells were then stained with DAPI for DNA (blue) and 3C antibody (red) as indicated. b DEF cells were transfected with pcDNA3.1-myc-His (−) A-3C and pcDNA3.1-myc-His (−) A for 24 h. Then DEF cells were stained with DAPI for DNA (blue) and 3C antibody (green) as indicated
Discussion
Many picornaviruses are significant human and animal pathogens that cause considerable economic burdens. Viral proteases (2A protease and 3C protease) have been attractive targets for the development of antiviral drugs [44, 45]. Proteases in the Caliciviridae family (3C-like protease, 3CL protease) and Coronaviridae family (3CL protease, papain-like protease) were shown to function similarly in proteolytic processing [46, 47]. Conserved viral genes, including those encoding a 3C protease or 3CL protease, characterize the viruses in the picornavirus-like supercluster. Possessing common characteristics, for example, conserved active sites, the 3C proteases and 3CL proteases serve as conducive targets for the design of broad-spectrum and safe antiviral drugs [48,49,50].
Sequence alignment suggests that the DHAV 3C protein possesses a 3C cysteine protease domain and a typical Cys-His-Asp catalytic triad. In this study, we applied FRET-based assays to determine the activity of DHAV 3C protease. When the peptide bond was cleaved by the 3C protease, it caused the separation of the fluorescence quencher moiety and fluorophore. Due to the disappearance of the FRET effect, the fluorescence of the fluorophore was detected. Hence, FRET-based assays have been applied successfully to monitor protease activity, and substrates that are recognized and cleaved are required for this assay. By testing autoprocessing sites (VP2-VP3, VP3-VP1, 2A-2C, 3A-3B, 3B-3C and 3C-3D), EV71 showed the most efficient enzyme activity towards the 3B-3C junction site. The EV71 3C protease showed the activity (Km = 30 ± 2 μM, Vmax = 85 ± 1.4 nM min− 1) against the 3B-3C junction site [38]. The 2C-3A junction could be rapidly hydrolyzed in poliovirus (PV)-infected HeLa cells. Based on the HPLC assay, the 2C-3A junction was the most efficient substrate for 3C protease of EV71 and CVA16. Subsequently, using FRET peptides confirmed that peptide containing 2A-3C junction was most efficiently cleaved with a Km of 63.2 ± 3.6 μM [44]. Therefore, the 2C-3A junction in the polyprotein of DHAV was used to design the fluorescence peptide. The DHAV 3C protease demonstrated the activity against the 2C-3A junction (Km = 50.78 μM, Vmax = 16.52 nmol/min). Here, we have report for the first time the substrate specificity and kinetic parameters of the DHAV 3C protease, which could provide basic information for antiviral development in the future. In addition, we have evaluated the inhibitory activity of AG7088 for the DHAV 3C protease. Further experiments are needed to detect other efficient substrates.
Our study shows that the DHAV 3C protease could localize to the nucleus of DEF cells, presumably due to the degradation of specific nucleoporins. This result is consistent with the observations of the 3C protease in other picornaviruses. The wild-type (WT) 3C protease sequence was altered with a histidine- or cysteine-to-alanine base substitution to create 3C protease mutants with greatly reduced catalytic activity. We observed fusion protease distribution throughout the cell except for some fluorescent signals in the cytoplasm of cells transfected with EGFP-3C-H38A and EGFP-3C-C144A. It was reported that when HeLa cells were transfected with EGFP-3C [containing a PV 3C gene sequence], EGFP expression was detected throughout the entire cells. Due to the generation of high quantities of free GFP in cells transfected with EGFP-3C, whether the 3C protease could localize to the nucleus was inconclusive [51]. In a previous report, it was confirmed that 3D and 3CD precursor proteins could enter the nucleus in PV-infected cells. However, these proteins localized to the cytoplasm of uninfected cells [51]. Furthermore, 2Apro of PV was demonstrated to be responsible for the redistribution of 3CD to the nucleus [52]. Subsequently, the HRV 3C protease was demonstrated to have a nuclear-targeting ability by causing the degradation of nuclear pore components [53]. The localization of the 3C protease in the nucleus is important for host cell transcription shut-off induced by picornaviruses. Hence, this has significant implications for future research.
The 3C protease is one of the most attractive viral proteins involved in the virus-host interaction and serves as a significant target for designing anti-picornavirus drugs [54]. More research on the protease activity of the viral 3C protease is still ongoing. For example, the 69th residue (Asp) of EV 3C has been identified as a novel and important site that is involved in protease activity and is a virulence determinant [55]. Furthermore, the 3C protease of PV lost protease activity when the 70th residue (Leu) was mutated to proline [56]. In this study, the 3C protease was obtained by prokaryotic expression, and its enzymatic activity was detected with a cleavage assay in vitro. Our results demonstrated the localization of the 3C protease in DHAV infected cells and transfected cells and its ability to move into the nucleus. These findings provide us with an important starting point to determine the function of the DHAV 3C protease.
Conclusion
We expressed, purified and evaluated the protease activity of the DHAV 3C protease for the first time. Similar to other picornaviral proteases, the activity of the DHAV 3C protease is temperature-, pH- and NaCl concentration-dependent. The kinetic analysis was calculated, and the Vmax and Km values were determined to be 16.52 nmol/min and 50.78 μM, respectively. Based on the FRET substrate-based assay, rupintrivir was found to exhibit inhibitory activity against the DHAV 3C protease. The DHAV 3C protease localizes throughout DHAV-infected cells and can enter into the nucleus without the cooperation of other viral proteins.
Abbreviations
- AIG1:
-
AvrRpt2-induced gene 1
- CREB-1:
-
CAMP response element-binding protein-1
- Dabcyl:
-
p-(p-dimethylaminophenylazo) benzoic acid
- DEF:
-
Duck embryo fibroblasts
- DHAV:
-
Duck hepatitis A virus
- Edans:
-
5′-[(2-aminoethyl) amino] naphthalene-1-sulfonic acid
- FRET:
-
Fluorescence resonance energy transfer
- HPLC:
-
High-performance liquid chromatography
- HRV:
-
Human rhinovirus
- ICTV:
-
International Committee on Taxonomy of Viruses
- IFA:
-
Indirect immunofluorescence assay
- IPTG:
-
Isopropyl thiogalactoside
- IRF7:
-
Interferon regulatory factor 7
- MAVS:
-
Mitochondrial antiviral signaling protein
- ORF:
-
Open reading frame
- RdRp:
-
RNA-dependent RNA polymerase
- RT-PCR:
-
Reverse transcription-PCR
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TAK1:
-
Transforming growth factor-β-activated kinase 1
- TBP:
-
TATA box binding protein
- TLR7:
-
Toll-like receptor 7
- TRIF:
-
TIR-domain-containing adapter-inducing interferon-β
References
Ou X, Mao S, Cao J, Cheng A, Wang M, Zhu D, Chen S, Jia R, Liu M, Sun K. Comparative analysis of virus-host interactions caused by a virulent and an attenuated duck hepatitis a virus genotype 1. PLoS One. 2017;12(6):e0178993.
Zhang R, Chen J, Zhang J, Yang Y, Li P, Lan J, Xie Z, Jiang S. Novel duck hepatitis a virus type 1 isolates from adult ducks showing egg drop syndrome. Vet Microbiol. 2018;221:33–7.
Levine P, Fabricant J. A hitherto-undescribed virus disease of ducks in North America. Cornell Veterinarian. 1950;40:71–86.
Swayne DE, Glisson JR. Diseases of poultry. (13th Edition), 2013.
Cha SY, Roh JH, Kang M, Kim B, Jang HK. Isolation and characterization of a low pathogenic duck hepatitis a virus 3 from South Korea. Vet Microbiol. 2013;162(1):254–8.
Chen LL, Xu Q, Zhang RH, Yang L, Li JX, Xie ZJ, Zhu YL, Jiang SJ, Si XK. Improved duplex RT-PCR assay for differential diagnosis of mixed infection of duck hepatitis a virus type 1 and type 3 in ducklings. J Virol Methods. 2013;192(1–2):12–7.
Hess JC, Paré JA. Viruses of waterfowl. In Seminars in avian and exotic pet medicine. 2004;13(4):176–83.
Wang W, Said A, Wang B, Qu G, Xu Q, Liu B, Shen Z. Establishment and evaluation of the goose embryo epithelial (GEE) cell line as a new model for propagation of avian viruses. PLoS One. 2018;13(3):e0193876.
Yugo DM, Hauck R, Shivaprasad HL, Meng XJ. Hepatitis virus infections in poultry. Avian Dis. 2016;60(3):576–88.
Wen X, Cheng A, Wang M, Jia R, Zhu D, Chen S, Liu M, Liu F, Chen X. Detection, differentiation, and VP1 sequencing of duck hepatitis a virus type 1 and type 3 by a 1-step duplex reverse-transcription PCR assay. Poult Sci. 2014;93(9):2184–92.
Shen Y, Cheng A, Wang M, Chen S, Jia R, Zhu D, Liu M, Sun K, Yang Q, Chen X. Development of an indirect ELISA method based on the VP3 protein of duck hepatitis a virus type 1 (DHAV-1) for dual detection of DHAV-1 and DHAV-3 antibodies. J Virol Methods. 2015;225:30–4.
Hu Q, Zhu D, Ma G, Cheng A, Wang M, Chen S, Jia R, Liu M, Sun K, Yang Q. A one-step duplex rRT-PCR assay for the simultaneous detection of duck hepatitis a virus genotypes 1 and 3. J Virol Methods. 2016;236:207–14.
Mao S, Ou X, Zhu D, Chen S, Ma G, Wang M, Jia R, Liu M, Sun K, Yang Q. Development and evaluation of indirect ELISAs for the detection of IgG, IgM and IgA1 against duck hepatitis a virus 1. J Virol Methods. 2016;237:79–85.
Cheng A, Wang M, Xin H, Zhu D, Li X, Chen H, Jia R, Yang M. Development and application of a reverse transcriptase polymerase chain reaction to detect Chinese isolates of duck hepatitis virus type 1. J Microbiol Methods. 2009;76(1):1–5.
Adams MJ, Lefkowitz EJ, King AMQ, Harrach B, Harrison RL, Knowles NJ, Kropinski AM, Krupovic M, Kuhn JH, Mushegian AR. Changes to taxonomy and the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses. Arch Virol. 2017;162(8):2505–38.
Ding C, Zhang D. Molecular analysis of duck hepatitis virus type 1. Virology. 2007;361(1):9–17.
Zhang Y, Cao Q, Wang M, Jia R, Chen S, Zhu D, Liu M, Sun K, Yang Q, Wu Y. The 3D protein of duck hepatitis a virus type 1 binds to a viral genomic 3′ UTR and shows RNA-dependent RNA polymerase activity. Virus Genes. 2017;53(6):831–9.
Spear A, Ogram SA, Morasco BJ, Smerage LE, Flanegan JB. Viral precursor protein P3 and its processed products perform discrete and essential functions in the poliovirus RNA replication complex. Virology. 2015;485:492–501.
Wen X, Cheng A, Wang M, Jia R, Zhu D, Chen S, Liu M, Sun K, Yang Q, Wu Y. Recent advances from studies on the role of structural proteins in enterovirus infection. Future Microbiol. 2015;10(9):1529–42.
Webber SE, Marakovits JT, Dragovich PS, Prins TJ, Zhou R, Fuhrman SA, Patick AK, Matthews DA, Lee CA, Srinivasan B. Design and synthesis of irreversible depsipeptidyl human rhinovirus 3C protease inhibitors. Bioorg Med Chem Lett. 2001;11(20):2683–6.
Lall MS, Jain RP, Vederas JC. Inhibitors of 3C cysteine proteinases from picornaviridae. Curr Top Med Chem. 2004;4(12):1239–53.
Wang HM, Liang PH. Picornaviral 3C protease inhibitors and the dual 3C protease/coronaviral 3C-like protease inhibitors. Expert Opin Ther Pat. 2010;20(1):59–71.
Lin YJ, Chang YC, Hsiao NW, Hsieh JL, Wang CY, Kung SH, Tsai FJ, Lan YC, Lin CW. Fisetin and rutin as 3C protease inhibitors of enterovirus A71. J Virol Methods. 2012;182(1–2):93–8.
Cao J, Ou X, Zhu D, Ma G, Cheng A, Wang M, Chen S, Jia R, Liu M, Sun K. The 2A2 protein of duck hepatitis a virus type 1 induces apoptosis in primary cell culture. Virus Genes. 2016;52(6):780–8.
Yang X, Cheng AC, Wang M, Jia R, Sun K, Pan K, Yang Q, Wu Y, Zhu D, Chen S. Structures and corresponding functions of five types of Picornaviral 2A proteins. Front Microbiol. 2017;8:1373.
Castello A, Alvarez E, Carrasco L. The multifaceted poliovirus 2A protease: regulation of gene expression by picornavirus proteases. J Biomed Biotechnol. 2011;20:369648.
Yalamanchili P, Datta U, Dasgupta A. Inhibition of host cell transcription by poliovirus: cleavage of transcription factor CREB by poliovirus-encoded protease 3Cpro. J Virol. 1997;71(2):1220–6.
Weng KF, Li ML, Hung CT, Shih SR. Enterovirus 71 3C protease cleaves a novel target Cstf-64 and inhibits cellular polyadenylation. PLoS Pathog. 2009;5(9):e1000593.
Kundu P, Raychaudhuri S, Tsai W, Dasgupta A. Shutoff of RNA polymerase II transcription by poliovirus involves 3C protease-mediated cleavage of the TATA-binding protein at an alternative site: incomplete shutoff of transcription interferes with efficient viral replication. J Virol. 2005;79(15):9702–13.
Lei X, Sun Z, Liu X, Jin Q, He B, Wang J. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by toll-like receptor 3. J Virol. 2011;85(17):8811–8.
Lei X, Xiao X, Xue Q, Jin Q, He B, Wang J. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J Virol. 2013;87(3):1690–8.
Lei X, Han N, Xiao X, Jin Q, He B, Wang J. Enterovirus 71 3C inhibits cytokine expression through cleavage of the TAK1/TAB1/TAB2/TAB3 complex. J Virol. 2014;88(17):9830–41.
Mukherjee A, Morosky SA, Delorme-Axford E, Dybdahl-Sissoko N, Oberste MS, Wang T, Coyne CB. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011;7(3):e1001311.
Ou X, Mao S, Cao J, Ma Y, Ma G, Cheng A, Wang M, Zhu D, Chen S, Jia R. The neglected avian hepatotropic virus induces acute and chronic hepatitis in ducks: an alternative model for hepatology. Oncotarget. 2017;8(47):81838.
Xie J, Wang M, Cheng A, Zhao XX, Liu M, Zhu D, Chen S, Jia R, Yang Q, Wu Y. Cytokine storms are primarily responsible for the rapid death of ducklings infected with duck hepatitis a virus type 1. Sci Rep. 2018;8(1):6596.
Wen X, Zhu D, Cheng A, Wang M, Chen S, Jia R, Liu M, Sun K, Zhao X, Yang Q. Molecular epidemiology of duck hepatitis a virus types 1 and 3 in China, 2010-2015. Transbound & Emerg Dis. 2018;65(1):10–5.
Mao S, Wang M, Ou X, Sun D, Cheng A, Zhu D, Chen S, Jia R, Liu M, Sun K. Virologic and immunologic characteristics in mature ducks with acute duck hepatitis a virus 1 infection. Front Immunol. 2017;8:1574.
Cui S, Wang J, Fan T, Qin B, Guo L, Lei X, Wang J, Wang M, Jin Q. Crystal structure of human enterovirus 71 3C protease. J Mol Biol. 2011;408(3):449–61.
Kuo C, Shie J, Fang J, Yen G, Hsu J, Liu H, Tseng S, Chang S, Lee C, Shih S, Liang P. Design, synthesis, and evaluation of 3C protease inhibitors as anti-enterovirus 71 agents. Bioorg Med Chem. 2008;16(15):7388–98.
Sweeney TR, Roqué-Rosell N, Birtley JR, Leatherbarrow RJ, Curry S. Structural and mutagenic analysis of foot-and-mouth disease virus 3C protease reveals the role of the β-ribbon in proteolysis. J Virol. 2007;81(1):115–24.
Sun D, Wang M, Wen X, Cheng A, Jia R, Sun K, Yang Q, Wu Y, Zhu D, Chen S. Cleavage of poly(a)-binding protein by duck hepatitis a virus 3C protease. Sci Rep. 2017;7(1):16261.
Boros Á, Pankovics P, Reuter G. Avian picornaviruses: molecular evolution, genome diversity and unusual genome features of a rapidly expanding group of viruses in birds. Infect Genet Evol. 2014;1(28):151–66.
Zhang JH, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4(2):67–73.
Shang L, Zhang S, Yang X, Sun J, Li L, Cui Z, He Q, Guo Y, Sun Y, Yin Z. Biochemical characterization of recombinant enterovirus 71 3C protease with fluorogenic model peptide substrates and development of a biochemical assay. Antimicrob Agents Chemother. 2015;59(4):1827–36.
Tan J, George S, Kusov Y, Perbandt M, Anemüller S, Mesters JR, Norder H, Coutard B, Lacroix C, Leyssen P. 3C protease of enterovirus 68: structure-based design of Michael acceptor inhibitors and their broad-spectrum antiviral effects against picornaviruses. J Virol. 2013;87(8):4339–51.
Blakeney SJ, Cahill A, Reilly PA. Processing of Norwalk virus nonstructural proteins by a 3C-like cysteine proteinase. Virology. 2003;308(2):216–24.
Akaji K, Konno H, Mitsui H, Teruya K, Shimamoto Y, Hattori Y, Ozaki T, Kusunoki M, Sanjoh A. Structure-based design, synthesis, and evaluation of peptide-mimetic SARS 3CL protease inhibitors. J Med Chem. 2011;54(23):7962–73.
Kuyumcu-Martinez NM, Van Eden ME, Younan P, Lloyd RE. Cleavage of poly (a)-binding protein by poliovirus 3C protease inhibits host cell translation: a novel mechanism for host translation shutoff. Mol Cell Biol. 2004;24(4):1779–90.
Dragovich PS, Prins TJ, Zhou R, Johnson TO, Hua Y, Luu HT, Sakata SK, Brown EL, Maldonado FC, Tuntland T. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 8. Pharmacological optimization of orally bioavailable 2-pyridone-containing peptidomimetics. J Med Chem. 2003;46(21):4572–85.
Mandadapu SR, Weerawarna PM, Prior AM, Uy RA, Aravapalli S, Alliston KR, Lushington GH, Kim Y, Hua DH, Chang KO. Macrocyclic inhibitors of 3C and 3C-like proteases of picornavirus, norovirus, and coronavirus. Bioorg Med Chem Lett. 2013;23(13):3709–12.
Sharma R, Raychaudhuri S, Dasgupta A. Nuclear entry of poliovirus protease-polymerase precursor 3CD: implications for host cell transcription shut-off. Virology. 2004;320(2):195–205.
Tian W, Cui Z, Zhang Z, Wei H, Zhang XE. Poliovirus 2A (pro) induces the nucleic translocation of poliovirus 3CD and 3C′ proteins. Acta Biochim Biophys Sin. 2011;43(1):38–44.
Ghildyal R, Jordan B, Li D, Dagher H, Bardin PG, Gern JE, Jans DA. Rhinovirus 3C protease can localize in the nucleus and alter active and passive nucleocytoplasmic transport. J Virol. 2009;83(14):7349–52.
Reich SH, Johnson T, Wallace MB, Kephart SE, Fuhrman SA, Worland ST, Matthews DA, Hendrickson TF, Chan F, Meador J. Substituted benzamide inhibitors of human rhinovirus 3C protease: structure-based design, synthesis, and biological evaluation. J Med Chem. 2000;43(9):1670–83.
Li B, Yue Y, Zhang Y, Yuan Z, Li P, Song N, Lin W, Liu Y, Gu L, Meng H. A novel enterovirus 71 (EV71) virulence determinant: the 69th residue of 3C protease modulates pathogenicity. Front Cell Infect Microbiol. 2017;7:26.
Uma M, Hegde SR, Rao PP, Nagalekshmi K, Gauthami S, Kumar D, Hegde NR. A novel point mutation (L70P) inactivates poliovirus 3C protease. Acta Virol. 2018;62(1):68–77.
Acknowledgments
We would like to thank many staff members who have helped with this paper. In addition, we thank AJE editing company for editing this manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (31472223), China Agricultural Research System (CARS-42-17), Sichuan Veterinary Medicine and Drug Innovation Group of China Agricultural Research System (CARS-SVDIP) and the Special Fund for the Key Laboratory of Animal Disease and Human Health of Sichuan Province (2016JPT0004).
Availability of data and materials
The datasets analyzed in this study are available from the corresponding author upon reasonable request.
Author information
Authors and Affiliations
Contributions
DS and MW designed and performed the experiments, DS analyzed the data and wrote the paper. XW, SM and AC conceived and designed the experiments. RJ, QY, YW, DZ, SC, ML, XZ, SZ, XC, YL, YY and LZ helped with the experiments. All authors read and approved the final manuscript for publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Animal Ethics Committee of Sichuan Agricultural University (2016–17).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sun, D., Wang, M., Wen, X. et al. Biochemical characterization of recombinant Avihepatovirus 3C protease and its localization. Virol J 16, 54 (2019). https://doi.org/10.1186/s12985-019-1155-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-019-1155-3