
Abstract
There are an estimated 400 million chronic carriers of HBV worldwide; between 15 and 20 million have serological evidence of exposure to HDV. Traditionally, regions with high rates of endemicity are central and northern Africa, the Amazon Basin, eastern Europe and the Mediterranean, the Middle East and parts of Asia. There are two types of HDV/HBV infection which are differentiated by the previous status infection by HBV for the individual. Individuals with acute HBV infection contaminated by HDV is an HDV/HBV co-infection, while individuals with chronic HBV infection contaminated by HDV represent an HDV/HBV super-infection. The appropriate treatment for chronic hepatitis delta is still widely discussed since it does not have an effective drug. Alpha interferon is currently the only licensed therapy for the treatment of chronic hepatitis D. The most widely used drug is pegylated interferon but only approximately 25% of patients maintain a sustained viral response after 1 year of treatment. The best marker of therapeutic success would be the clearance of HBsAg, but this data is rare in clinical practice. Therefore, the best way to predict a sustained virologic response is the maintenance of undetectable HDV RNA levels.
Similar content being viewed by others


Background
The hepatitis delta or D virus (HDV) was discovered in 1977 by Rizzetto et al. during an analysis of liver biopsies from patients chronically infected with the hepatitis B virus (HBV) and more severe liver damage [1]. Analysis of the hepatocytes using the technique immunofluorescence showed the presence of a specific antigen, which was named with the Greek letter δ and subsequently renamed the Delta antigen. Concomitantly, the authors also found specific antibodies against the delta antigen (anti-HDAg) in the serum of these patients [1]. Initially, they believed it was a new serological marker for HBV; however, studies with chimpanzees made the association between HBV and HDAg even clearer [2].
Later it was demonstrated that HBV infection associated with HDAg (HBV/HDV) did not develop in chimpanzees that previously presented titers of antibodies against HBsAg (anti-HBs). However, the rapid rise and persistence of HDAg was observed in chimpanzees who were chronically infected with HBV. It was suggested that HDAg could be a new marker of a transmissible pathogen, an HBV variant or even another viral agent that would need HBV’s helper functions [3]. In their seroprevalence study, Rizzetto et al. noted the presence of anti-HDAg in the serum of patients from Italy and from around the world, suggesting that HDAg was widely distributed [4,5,6].
The next discovery was that, besides the association of this new antigen with HBsAg, HDAg presented itself as complexed to a small RNA [7], showing the possible genetic material of these viruses, demonstrating that their genetic material was not dependent on the HBV genome. Therefore, HDV was known for containing the smallest genome among the known RNA viruses (approximately 1682 base pairs), resembling viruses that infect plants [8]. In 1986 HDV was described as the first animal virus identified with a circular RNA genome due to the similarity of nucleotides; circular RNAs had hitherto only been found in plant viruses. It was noted that the size and its secondary structures were similar to higher plant viroid and virusoid RNA, which suggested that HDV may have originated from the plant instead of the animal world [9]. Another unique feature of RNA HDV is the ability to self-cleave due to the presence of a ribozyme in the genomic and antigenomic sequence of its RNA, discovered in 1989, which is a sequence of about 85 nucleotides capable of self-cleavage and self-binding [10]. Ribozymes are also present in viroids, although ribozymes described from plant viruses were different from those found in HDV [11].
Despite the presence of the ribozyme, research continued to emerge about how HDV could replicate with a very small genome encoding only HDAg. It was assumed that HDV used HBV’s machinery and cellular functions to enable its replication [12]. Only in 1993 was an important study published by Taylor and Fu [13] for understanding the role of HBV and hepatocytes in the replication of HDV. Transcription of genomic into antigenomic viral RNA was observed along with the fact that this process was carried out by the host cells’ RNA polymerase II. It was believed that the rod shape of the viral RNA would be recognized as double stranded DNA by RNA Polymerase II [13,14,15]. The final replication step is the rearrangement of the genomic and antigenomic RNA of HDV which is catalyzed by its ribozymes, which cleave the product of the genome/antigenome transcription to form the circular structure of HDV RNA [13, 16].
Classification
HDV was recognized by the International Committee on Taxonomy of Viruses (ICTV), as a new virus species from vertebrates, the only representative of the Deltaviridae family, Deltavirus genus [17]. Although HDV is quite similar structurally and in its mode of replication to phytopathogens, viroids and virusoids, it is sufficiently different to be assigned to a separate genus. HDV is commonly classified as a satellite virus of HBV, as it is based on the biological principle that HDV is incapable of infection in the absence of HBV [14, 18].
Viral structure
HDV is formed, on the outside, by a spherical lipoprotein envelope containing HBsAg [19]. On the inside of the virion there is a ribonucleoprotein composed of the viral genome complexed to HDAg (Fig. 1) [8, 18, 20].

Schematic representation of HDV. The Hepatitis D virus wrapped in the HBV surface antigen. Outer HBsAg in black. With L (preS1 or large), M (preS2 or middle) and S (small) peripheral distribution of the three surface proteins. In the central portion, HDAg (HDAg-S and HDAg-L) is represented in blue and single-stranded RNA in red
The viral genome consists of one single-stranded circular RNA (ssRNA) of about 1.7 kb and negative polarity. Due to the large amount of GC, nearly 74% of the nucleotides complement themselves intramolecularly, may fold into a secondary structure similar to a rod and are found complexed to HDAg [14, 18, 21, 22].
Delta antigen
The Delta (HDAg) antigen is the only protein encoded by HDV, a phosphoprotein and can be found in two protein forms: a short one named HDAg-S and a large one called HDAg-L, with molecular weights of 24 kilodaltons (195 amino acids) and 27 kilodaltons (214 amino acids), respectively [23]. Studies have shown that HDAg-S promotes RNA replication while HDAg-L promotes HDV RNA enveloping to assemble the virion [21, 24,25,26,27].
During the replication cycle, the antigenome undergoes a post-transcriptional modification where the gene encoding HDAg-S is modified by an enzyme called Adenosine Deaminase (ADAR1), a host protein, substituting an adenine for an inosine, indirectly exchanging the UAG-stop codon for a UGG-tryptophan, known as the Amber/W site, which will give rise to the gene encoding HDAg-L with an additional 19 amino acids (Fig. 2) [28,29,30,31].

HDV RNA editing mechanism. Genomic RNA (gray rounded rectangle) serves as a template for mRNA synthesis which is translated into HDAg-S, which is necessary for synthesis of new RNAs. The genome serves as a template for the synthesis of the antigenome (rounded black rectangle), which also serves as a template for new genomic RNAs. A fraction of antigenomic RNAs are edited by the enzyme adenosine deaminase (ADAR-1) at the UAG site (stop codon), also called the Amber/W site (wavy and dotted arrow), wherein an adenine is replaced by inosine. The edited antigenomes serve as templates for the edited genomes (rounded rectangle edited for ACC). Edited genomes serve to synthesize edited messenger RNAs (with UGG – Tryptophan instead of UAG-stop codon) encoding HDAg-L, which is the key factor for enveloping the virus and inhibiting replication. The edited genome and antigenomes are simultaneously synthesized by a replication mechanism called rolling-circle. This way the editing levels accumulate replication products. Note that the numbering scheme is intended to indicate an increasing repertoire of activities that persist as replication occurs, rather than a gradual progression in which the above processes are terminated. (Source: [28]. Curr Top MicrobiolImmunol, our translation, modified)
The difference between the two forms of HDAg is in the 19 additional amino acids in the C-terminal region of HDAg-L. Both HDAg isoforms have multiple functional domains in common, including the RNA-binding domain (RBD), a nuclear localization signal (NLS) a coiled coil domain (CCD) and a C-terminal portion of the sequence rich in proline and glycine. The 19 additional amino acids of HDAg-L are a virus assembly signal (VAS), which is a highly variable and specific sequence for each genotype [32]. Central to virus assembly, it serves as a binding site interacting with HBsAg/membrane (Fig. 3) [14].

Structures of the two types of Delta antigen. (Source: [14], Journal of Virology, modified)
HDAg undergoes several other post-transcriptional modifications such as phosphorylation [33, 34], acetylation [35] and methylation [36] and in the case of HDAg-L, isoprenylation [37]. The methylation of Arg13 [36, 38] acetylation of Lys72 [39], and phosphorylation of Ser177 [40] and Ser123 [41] were related to the subcellular localization of HDAg and RNA replication. Most of these changes are important for the functions of HDAg-S in HDV RNA replication which acts by directly stimulating the elongation of transcription through the substitution of the transcription repressor elongation factor linked to RNA polymerase II [42, 43].
HDV RNA
Replication of the genome is completely directed by RNA, i.e., all the synthesis of new RNA has as its template HDV RNA itself, with no intermediate DNA template in the replication. In hepatocytes, HDV synthesizes complementary RNA, called antigenome, from its genome (Fig. 4) [32].

Schematic representation of HDV RNAs. Antigenome and genome of approximately 1700 nucleotides in a circular conformation (black). HDAg-S with 195 amino acids (orange) and HDAg-L with 214 amino acids (orange + blue). Negative polarity, the genome has an inverse orientation from the messenger RNA
The genome and antigenome contain one ribozyme domain each with about 85 nucleotides [44, 45] which have the capacity to self-cleave and self-bind; this activity is an absolute requirement for viral RNA replication [46, 47].
Viral replication
HDV uses a replication pathway called double-rolling circle, which is very similar to the strategy used by viroids, virusoids, and viroid-like satellite RNAs [48]. In general, the main feature of this type of replication is the use of a circular RNA strand as a template, which is transcribed by an RNA-dependent RNA polymerase of the host or helper virus. It is noteworthy that HDV is the only human pathogen that uses the host enzyme [49,50,51].
However, in the case of HDV where the host cell is a hepatocyte and due to the inexistence of RNA-dependent RNA polymerase in eukaryotic cells, in this replication the virus is deceptive and uses the hepatocyte’s own RNA polymerase. Subsequently, the new RNA strands suffer ribozyme-catalyzed cleavage and, finally, are connected by host cell enzymes (Fig. 5) [52].

Replication and assembly cycle diagram. (1) The virion adheres to the hepatocytes via an interaction between HbsAg-L and a membrane receptor that has not yet been characterized, in the host cell, (2) the virion enters the cell and loses its envelope. (3) The ribonucleoprotein (HDV RNA complexed to HDAg) is imported into the nucleus of the cell, (4) the genomic RNA is transcribed in the nucleus into mRNA and antigenomic RNA, which in turn serves as a template for new RNA genomic transcripts. (5) The mRNA is exported to the cytoplasm where it is translated into HDAg-S in the endoplasmic reticulum, (6) the new HDAg-S molecules return to the cell nucleus to support the replication of more RNA. The two forms of HDAg associate themselves with the new genomic RNA to form new ribonucleoproteins, (7) which are exported to the cytoplasm where they will interact with HBV envelope proteins through HDAg-L in the endoplasmic reticulum to form new viral particles. (8) These particles by budding in an intermediate compartment (9) are exported from the hepatocyte via the trans-Golgi network to re-infect new cells. (Source: [20], Lancet, modified)
In recent decades several studies have contributed to understanding HBV replication [53, 54] and suggest that the same mechanisms are involved in the initial process of hepatocyte infection. Initially, the surface proteins derived from the HBV envelope, would bind to heparan sulfate proteoglycans (HSPG). This connection, despite having low affinity [55], is important to the infectious process, since it helps in adhesion of the virion to its cellular receptor, the Na+ taurocholate co-transporting polypeptide (NTCP) [56]. Only after this virion [57,58,59] binding step with NTCP, high affinity binding occurs which will start the viral particle entrance process by endocytosis-mediated internalization [54, 60].
Upon entry into the cell, the HDV ribonucleoprotein is released into the cytoplasm and translocated by HDAg to the cell nucleus via the nuclear localization site (NLS); the ribonucleoprotein is imported by cellular importins [57,58,59].
In the nucleus, specifically in the nucleoplasm, the genomic RNA is transcribed by RNA polymerase II in unmodified mRNA that, in turn, migrates to the cytoplasm where it is translated into HDAg-S (required for replication of the HDV RNA) [13, 15, 55, 61]. In the nucleus, more specifically in the nucleosome, the genomic RNA is transcribed by RNA polymerase I [62] in a complementary RNA template, called antigenomic RNA. In the nucleoplasm, antigenomic RNA is transcribed by RNA polymerase II into new genomic RNA [61].
Later a fraction of antigenomic RNA undergoes editing by ADAR1 to serve as a template for edited genomic RNA; hence, the mRNA is edited and subsequently will give rise to HDAg-L [28]. Studies have shown that errors in the addition to polymerase and ADAR1-catalyzed amber/W editing, RNA recombination is also known can contribute to the genetic heterogeneity of HDV [51, 63, 64]. Some study show that recombination is not rare [65]. In the study realized by Sy et al. (2015), was show that can occur in the HDV genome for recombination located to the position nt908 in the genome which is downstream of the ribozyme activity (Rz) and polyadenylation signal sequence [66]. This recombination can contribuit for variability the genotypes HDV.
The two HDAg isoforms are sent on to the nucleus where they will be associated with the new unedited genomic RNA, forming a new ribonucleoprotein, which is exported to the cytoplasm [58]. Thus, it was observed clearly demonstrated that HDV RNA can replicate in hepatocytes and form HDV RNPs without the assistance of HBV [67]. However, HBV is indispensable for a productive HDV infection, considering that, in the assembly of the virion, HDAg-L will interact with HBsAg in the endoplasmic reticulum for viral involvement, thus producing an infectious viral particle, where, after there is the release of new viral particles through the Golgi complex, will be able to infect other cells [58, 68].
Genotypes and epidemiology
The viral genetic diversity is related to the geographic origin of the isolates, and so far eight genotypes have been identified classified as HDV-1 through HDV-8 [69,70,71,72].
There are an estimated 400 million chronic carriers of HBV worldwide, between 15 to 20 million have serological evidence of exposure to HDV [72, 73]. Traditionally, regions with high rates of endemicity are central and northern Africa, the Amazon Basin, eastern Europe and the Mediterranean, the Middle East and parts of Asia [74]. HDV-1 is ubiquitous [75] and is often isolated in the United States, Europe and the Middle East, but has also been isolated in Russia, Africa, Asia and Brazil [24, 32, 71, 75,76,77,78] HDV-2, formerly known as genotype IIa, is found in Japan, Taiwan and Russia [79,80,81]. HDV-3 has been isolated in the Amazon region (Peru, Colombia, Ecuador and Brazil) [78, 82,83,84,85]. HDV-4 (the old genotype IIb) is found in Taiwan and Japan [80, 81, 86]. The genotypes HDV-5, HDV-6, HDV-7 and HDV-8 are found in Africa [69, 71]. HDV-8 was isolated in the countryside of the state of Maranhão (Brazil) in two native individuals [87].
HDV-3 is responsible for outbreaks of severe and common fulminant hepatitis in northeastern South America [76]. Studies show that HDV-3 is prevalent in the Brazilian Amazon. This genotype is apparently related to the more aggressive nature of HDV [76, 77, 84, 85, 88].
The western Amazon basin, including Brazil, Peru, Ecuador, Venezuela and Colombia, represents one of the highest rates of HBV infection in the world [89]. In Brazil, this area corresponds to the states of Acre, Amazonas, Rondônia and Roraima, with significant prevalence in the indigenous population [90, 91].
Braga et al. [92], in a study about the prevalence of HBV in Labrea, Amazonas, 11 years after the introduction of the HBV vaccine in the region, showed an HBsAg prevalence of 3.3% and an anti-HBc prevalence of 49.9%. However, in a study done previous to the introduction of the hepatitis B vaccine, the prevalence of HBsAg in this region of the Amazon was described as 16.7% [90]. Despite the prevalence of HBsAg having had a significant decline in the region, it is still considered a region with moderate endemicity for HBV.
In the state of Amazonas, the basins of the Juruá, Solimões and Purus rivers are areas of high endemicity for HBV and HDV, representing a major public health problem with serious cases, including fulminant hepatitis. A recent study in the region reported that almost a third (29.5%) of patients with HBV were co-infected with HDV [89]. Similar results were also described in Lábrea by Braga et al. [92]; of those who tested positive for HBsAg, the prevalence of anti-HDV was 30%. Later, Braga and colleagues described an anti-HDV prevalence of over 40% in chronic HBV carriers [93]. Fonseca obtained similar results, before the introduction of the hepatitis B vaccine, describing in the state of Amazonas an anti-HDV prevalence of 34.4% in patients chronically infected with HBV [90]. In Brazil, outside the Amazon region, few studies on the prevalence of HDV have been done, showing a very low prevalence.
In Brazil, studies show a significantly lower frequency in patients not co-infected with HIV than those co-infected with HIV, contrary to that described for other countries where prevalence can reach 50% in this population. A study about HDV prevalence in patients co-infected with HIV-HBV in the state of São Paulo, showed a rate of HDV of 1.2%, only one patient with anti-HDV was positive of the 81 with chronic hepatitis B. Similarly, in the state of Mato Grosso, in an analysis of 37 HBsAg positive patients only one was positive for HDV (2.7%). Corroborating data are described even in endemic areas, like in the Western Amazon, where a prevalence of 9.4% in patients with chronic hepatitis B was described. Although an increased prevalence was observed compared to the other two studies, it is significantly lower compared to the prevalence in the non-HIV population (34.4%) [94].
Clinical aspects
Due to the need for association with HBV, HDV can only be transmitted in the presence of a concomitant infection with HBV in one of two ways (super-infection or co-infection), depending on the previous status of the individual for HbsAg (Fig. 6) [95].

Schematic representation of the clinical course of hepatitis Delta
Co-infection is the simultaneous acute infection of HBV and HDV in a susceptible individual; this infection begins only after HBV has infected hepatocytes, and is similar to acute hepatitis B [96]. Because HBV is essential for HDV, the rate of progression to chronicity is similar to acute hepatitis B, i.e. between 2 and 5% [97, 98]. The incubation period of hepatitis D is dependent on HBV inoculum titers which determine the incubation time of hepatitis B [16]. Acute hepatitis may present itself as monophasic (one phase) and biphasic (two different phases), the first phase being dependent on HBV titers and the second on HDV titers [95]. In 95% of cases, spontaneous healing is seen, making it an important cause of severe or fulminant hepatitis [99].
Super-infection is an HDV infection in an individual chronically infected with HBV. It can cause fulminant hepatitis and chronicity rates are above 80%, which is related to an increased risk of early development of cirrhosis and hepatocellular carcinoma [1, 100, 101] It has a varied clinical appearance, although it generally causes a more severe acute hepatitis and has a relatively short period of incubation. In asymptomatic HBsAg patients it can lead to acute hepatitis, while in patients with chronic active hepatitis B, it can lead to exacerbation of symptoms, with liver decompensation. Patients who have a super-infection progress to chronic hepatitis in approximately 90% of cases [95].
HDV infection, whether in the form of a co-infection or a super-infection, is a significant cause of fulminant viral hepatitis, which is caused by HDV more often than by other forms of viral hepatitis [95]. A study conducted in Samara, Russia, showed that of the 27 diagnoses of fulminant viral hepatitis, 13 were infected with HDV, and of these 11 were males and 2 were females [102]. Some studies in different countries of South America show that acute hepatitis caused by an HBV and HDV co-infection or super-infection is the major cause of severe acute hepatitis in the Amazon region [84, 103].
According to a prospective analysis of 33 patients with chronic HDV admitted to a Spanish hospital in the period from 2006 to 2007, viral replication of HBV and HDV was quite variable during the study. In 54.5% HDV replication predominated, in 30.3% HBV predominated and in 15.2% HBV and HDV maintained similar levels. These data suggest that there is a suppression of HBV by HDV, but with important replication fluctuation of the two viruses [104]. The persistence of HBV replication, even at minimum levels, is associated with worse hepatocellular damage [105]. A third form has been described in patients after liver transplantation: latent infection [101]. Characterized by the presence of anti-HDV in the liver, hepatocyte nuclei in the absence of HBsAg and HDV RNA in the blood, is associated with low hepatocellular damage.
The chronic form of hepatitis D is the most severe and rapidly progressive of all chronic viral hepatitides. It leads to cirrhosis in approximately 70% within 5 to 10 years, usually involving patients of a young age. One to 2 years after the episode of acute hepatitis D, 15% develop cirrhosis. The risk of developing cirrhosis is 3 times higher in an HDV infection than in an HBV mono-infection [106]. A study in the Amazon region of Brazil showed that over 50% of treated patients treated with chronic hepatitis D who underwent a biopsy had moderate to intense fibrosis [91].
Fulminant hepatitis D has a dramatic evolution, with very poor prognosis. The clinical course occurs varying from 4 to 30 days after the onset of acute hepatitis D symptoms [107]. Transaminase levels may be high, but with massive liver necrosis, these levels tend to decrease rapidly. The same occurs with HDV replication levels, since there are few viable hepatocytes. If there is no liver transplantation in the first 10 days, mortality reaches approximately 80% [95].
Diagnosis
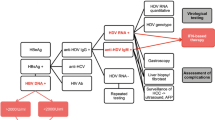
The first step in the diagnosis of HDV is screening for antibodies against HDAg (anti-HDVIgM and IgG) in HBsAg positive individuals. In patients with the anti-HDV reagent, the next step is to screen for HDV RNA in the serum to determine whether the presence of the antibody against HDAg reflects a persistent active infection (HDV RNA positive) or only represents a decreasing serological scar (HDV RNA negative). In individuals with an HDV infection and liver disease (changes in Alanine aminotransferase (ALT), see Fig. 7) it is crucial to distinguish the type of HDV/HBV infection, whether it be an acute co-infection or super-infection in patients with chronic HBsAg, since the prognosis and management of the two types of infection are different [108, 109].

Fluxogram for the diagnosis of hepatitis Delta
In individuals who have the reagent for HBsAg and live in areas endemic for hepatitis Delta (such as the western Amazon) it is recommended to determine the phase of HBV infection (acute or chronic) through screening for total anti-HBc and IgM. Acute HBV infection is characterized by the presence of anti-HBc IgM, and when accompanied by total anti-HDV and/or IgM reagents, it corresponds to an HDV co-infection. Chronic HBV infection is characterized by the presence of anti-HBc IgG (total anti-HBc reagent with non-reactive anti-HBc IgM), and when accompanied by total anti-HDV and/or IgM reagents it corresponds to an HDV super-infection (Fig. 7) [110, 111].
Both in an HDV co-infection and an HDV super-infection, screening for HDV RNA using molecular techniques such as polymerase chain reaction (PCR) is necessary, as the goal is to quantify the circulating virus in the blood (real time PCR) and determine the genotype (conventional PCR, restriction fragment length polymorphism - RFLP or sequencing) 76, [112],119. In the case that HDV RNA is negative it is recommended to repeat PCR and/or if necessary, the use of other diagnostic methods such as immunohistology for a liver biopsy and biochemical methods like verification of ALT. It is also important to screen for other viral infections such as the hepatitis C virus (HCV) by screening for anti-HCV and/or HCV RNA [113].
Liver biopsy is an important tool for the elucidation of the diagnosis of more than 20% of patients with unknown causes of cirrhosis [114]. Histologically acute hepatitis D is characterized by necrosis and hepatocellular inflammation, with lymphocytes and Kupffer cells infiltrating the parenchyma and portal region. This described pattern is not specific to hepatitis D, but despite being common to all forms of viral hepatitis, in hepatitis D it generally tends to be more intense [16]. Injury to the hepatocyte is typically focal, except in severe cases where confluent necrosis is seen [115]. In chronic hepatitis D, the histopathology consists of hepatocellular necrosis and inflammation in the parenchyma and portal region, associated with varying degrees of liver fibrosis [116]. Periportal necrosis is most commonly found in the other forms of viral hepatitis [95]. In patients with HDV, the definitive diagnosis by liver biopsy is done by identifying HDAg [114]. The amount of HDAg decreases with the progression of fibrosis, being almost undetectable in the final stage of the disease [16].
Treatment
The appropriate treatment for chronic hepatitis delta is still widely debated because of the lack of an effective drug. Alpha interferon is currently the only therapy licensed for the treatment of chronic hepatitis D [117, 118]. The most used drug is pegylated interferon, but only about 25% of patients maintain a sustained viral response after 1 year of treatment [119]. Transaminase levels normalize in only 40–70% of treated patients and relapse occurs in 60–97% [120]. A sustained virologic response is described as occurring when the viral load is negative 6 months after the end of treatment [119].
Some studies were developed in an attempt to find a more effective therapy than Interferon monotherapy. However, these studies were not encouraging showing that monotherapy with lamivudine, entecavir, adefovir, tenofovir, famciclovir and ribavirin or combination therapy with interferon and a nucleos (t) ide analog was no more effective than monotherapy with Interferon [121,122,123,124].
The duration of therapy is also controversial but some studies have shown that 2 years of treatment did not obtain better results than those treated for 1 year [125, 126]. Patients with HDV viremia and elevated transaminases should initiate treatment with Peg-interferon alpha 2a or 2b for 1 year. In cases where HDV RNA is negative, treatment should be terminated and the patient monitored. However, in cases where after 1 year of therapy there still remain detectable levels of HDV RNA, another year of therapy is indicated, especially with elevated transaminase levels [119].
In patients with decompensated cirrhosis, the use of interferon is contraindicated, but in patients with compensated cirrhosis, peg-interferon was effective [127].
Later studies suggest that the virologic response was not altered by previous treatment with interferon, with similar results being described in two major studies [125, 128], with virologic response rates sustained between 20 and 25%, both in naive patients as well as in those being re-treated.
Alternative treatment options are currently being explored in clinical trials, prenylation inhibitors are promising and has been assessed in patients with chronic HDV infection. Lonafarnib showed a dose-dependent reduction of HDV RNA levels in patients after 28 days of therapy [129].
With recent description of the receptor-mediated entry HBV, also used by HDV, novel therapeutic strategies for treatment of Delta hepatitis was developed that interfer this entry. Blockade of the HBV-HDV-specific receptor sodium/taurocholate cotransporting polypeptide NTCP by Myrcludex show promising results [130]. The HBV entry inhibitor myrcludex B is also being developed. Bogomolov and cols described a myrcludex B in patients with chronic hepatitis D infection either alone or in combination with Peg-INF alpha 2a as compared to patients only treated with Peg-INF alpha 2a [131]. Myrcludex B monotherapy was associated with HDV RNA and HBV DNA declines and improvement of ALT levels after 24 weeks of treatment. The antiviral effect was more pronounced en combination with Peg-INF alpha 2a [131].
Preliminary results of 6 months treatment with a subcutaneous HBV PreS1-derived myristoyled peptide as an entry inhibitor indicates an encouraging short-term response with low side-effects [132].
Likewise, the observation that HDV assembly requires farnesyltransferase, have enabled another novel therapeutic strategies. Short-term use of oral farnesyl transferase inhibitors induces a log10 reduction of viral RNA in almost all treated patients, but is associated with gastrointestinal upset and weight loss (especially using 200 mg/day) [133].
Recent studies comparing the antiviral effects of Peg-INF alpha, Peg-INF lambda and entecavir in human liver chimeric mice demonstrated that Peg-INF alpha and Peg-INF lambda reduced HDV viremia, serum HBsAg levels and intrahepatic levels of genomic and antigenomic HDV RNA [134].
Many treatment protocols have been tested worldwide (Table 1). Knowledge, Borzacov and colleagues [135] reported in a clinical study as better virologic response at the end of treatment (95%) and sustained virologic response (95%) for the treatment of delta hepatitis.
Therapeutic response markers
The best marker of therapeutic success would be the clearance of HBsAg, but this data is rare in clinical practice [136]. Therefore, the best way to predict the sustained virologic response is the maintenance of undetectable HDV RNA levels [119]. However, therapeutic success may be related to the decrease in HDV RNA and HBsAg levels and the normalization of ALT; even without its undetectability, it is associated with reduced events related to liver disease when treated with IFN [135, 137]. The treatment showed, in most studies, a low percentage of undetectability and a high rate of relapse, even if it was late. An early virological response, i.e., undetectable HDV RNA or a 3 log decrease during the first 6 months of treatment, may be indicative of a sustained virologic response [122, 139, 142].
Hepatocellular carcinoma
The increased frequency of cirrhosis associated with increased induction of liver inflammation in chronic carriers of HDV represent indirect risk factors for HCC [138]. However, a further increase in oncogenicity due to HDV infection itself has not been proven [139, 140].
Some studies suggest that an HBV/HDV co-infection increases the risk of HCC [140], showing a 3-fold increase in the risk of HCC and 2 times the mortality compared to patients with an HBV mono-infection [106, 141]. However, a retrospective analysis of 962 patients with HBV, 82 of whom were co-infected with HDV, showed similar rates of HCC in both groups [142]. Thus, this relationship is still controversial in the literature and the role of HDV in the induction and development of HCC should be further studied.
Prognosis
An Italian study conducted in Milan analyzed 299 patients with chronic hepatitis D admitted between 1978 and 2006; the probability of survival 20 years after the diagnosis of chronic HDV infection was 86%. The persistent replication of HDV was the only predictive factor associated with an increased risk of mortality [98]. However, quantitative values of HDV RNA do not seem to be correlated with the degree of liver disease in patients infected with HDV [143].
Farci et al., in a longer follow-up, on average 13 years, of patients previously treated with IFN alpha 2a 9MU (n = 14), 3MU (n = 14) and the control group (n = 14) showed that, although no patients had negated the viral load, survival was significantly higher in those who used high IFN doses, with no significant difference between those who used low doses and the controls. A significant improvement in necroinflammatory lesions was also observed in the liver biopsy of patients treated with high doses. In this study, there is an estimated survival in 12 years after treatment, without the need for transplantation, of 86% among those who received 9MU, 39% with 3MU and 31% in the control group [135].
A recent cohort study shows that anti-HDV IgM may persist in patients with chronic hepatitis from HDV or reappear in who have relapsed after treatment. It may be associated with increased levels of ALT and bilirubin and decreased levels of albumin, as well as histological activity [144].
Conclusion
This review of the clinical and virological aspects of hepatitis D demonstrates strategies for the diagnosis and treatment of the disease. Unfortunately, the lack of an effective anti-HDV drug directs therapy for pegylated interferon, but only a small percentage maintains the virological response after 1 year of treatment. Therefore, therapeutic success may be related to decreased levels of HDV and HBsAg and normalization of ALT, however, even without its undetectability, it is associated with reduced events related to liver disease when treated with pegylated interferon.
Abbreviations
- ALT:
-
Alanine aminotransferase
- anti-HDAg:
-
Antibody produced in our body to fight the antigen (HDAg)
- CCD:
-
Coiled coil domain
- HBsAg (anti-HBs):
-
Antibody produced in our body to fight the antigen (HBsAg)
- HBsAg:
-
Hepatitis B virus surface antigen
- HBV:
-
Hepatitis B virus
- HCC:
-
Hepatocellular carcinoma
- HCV:
-
Hepatitis C virus
- HDAg:
-
Antigen Delta
- HDAg-L:
-
Protein large antigen Delta
- HDAg-S:
-
Protein short antigen Delta
- HDV:
-
Hepatitis D virus
- HIV:
-
Human immunodeficiency virus
- HSPG:
-
Heparan sulfate proteoglycans
- ICTV:
-
International Committee on Taxonomy of Viruses
- NLS:
-
Nuclear localization signal
- NLS:
-
Nuclear localization site
- NTCP:
-
Na+ taurocholate co-transporting polypeptide
- PCR:
-
Reaction chain polymerase
- RBD:
-
RNA-binding domain
- RFLP:
-
Restriction fragment length polymorphism
- RNA:
-
Ribonucleic acid
- ssRNA:
-
Single-stranded circular RNA
References
Rizzetto M, Canese MG, Aricò S, Crivelli O, Trepo C, Bonino F, et al. Immunofluorescence detection of new antigen-antibody system (delta/anti-delta) associated to hepatitis B virus in liver and in serum of HBsAg carriers. Gut. [Internet]. 1977;18:997–1003. Available from: http://www.ncbi.nlm.nih.gov/pubmed/75123. Cited 29 Mar 2016.
Canese MG, Rizzetto M, Arico S, Crivelli O, Zanetti AR, Macchiorlatti E, et al. An ultrastructural and immunohistochemical study on the delta antigen associated with the hepatitis B virus. J Pathol. [Internet]. 1979;128:169–75. Available from: http://www.ncbi.nlm.nih.gov/pubmed/392063. Cited 29 Mar 2016.
Rizzetto M, Canese MG, Gerin JL, London WT, Sly DL, Purcell RH. Transmission of the hepatitis B virus-associated delta antigen to chimpanzees. J Infect Dis. [Internet]. 1980;141:590–602. Available from: http://www.ncbi.nlm.nih.gov/pubmed/6989929. Cited 29 Mar 2016.
Rizzetto M, Purcell RH, Gerin JL. Epidemiology of HBV-associated delta agent: geographical distribution of anti-delta and prevalence in polytransfused HBsAg carriers. Lancet (London, England). [Internet]. 1980;1:1215–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/6104036. Cited 29 Mar 2016.
Rizzetto M, Shih JW, Gerin JL. The hepatitis B virus-associated delta antigen: isolation from liver, development of solid-phase radioimmunoassays for delta antigen and anti-delta and partial characterization of delta antigen. J Immunol. [Internet]. 1980;125:318–24. Available from: http://www.ncbi.nlm.nih.gov/pubmed/6769999. Cited 29 Mar 2016.
Smedile A, Rizzetto M. HDV: thirty years later. Dig Liver Dis. [Internet]. 2011;43(Suppl 1):S15–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21195366.
Bonino F, Hoyer B, Ford E, Shih JW, Purcell RH, Gerin JL. The delta agent: HBsAg particles with delta antigen and RNA in the serum of an HBV carrier. Hepatology [Internet]. 1:127–31. Available from: http://www.ncbi.nlm.nih.gov/pubmed/6169614. Cited 29 Mar 2016.
Rizzetto M, Hoyer B, Canese MG, Shih JW, Purcell RH, Gerin JL. Delta agent: association of delta antigen with hepatitis B surface antigen and RNA in serum of delta-infected chimpanzees. Proc Natl Acad Sci USA. [Internet]. 1980;77:6124–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/6934539. Cited 6 June 2016.
Kos A, Dijkema R, Arnberg AC, van der Meide PH, Schellekens H. The hepatitis delta (delta) virus possesses a circular RNA. Nature. [Internet]. 1986;323:558–60. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2429192. Cited 6 June 2016.
Wu HN, Lin YJ, Lin FP, Makino S, Chang MF, Lai MM. Human hepatitis delta virus RNA subfragments contain an autocleavage activity. Proc Natl Acad Sci U S A. [Internet]. 1989;86:1831–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2648383. Cited 6 June 2016.
Salehi-Ashtiani K, Lupták A, Litovchick A, Szostak JW. A genomewide search for ribozymes reveals an HDV-like sequence in the human CPEB3 gene. Science. [Internet]. 2006;313:1788–92. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16990549. Cited 6 June 2016.
Branch AD, Benenfeld BJ, Baroudy BM, Wells FV, Gerin JL, Robertson HD. An ultraviolet-sensitive RNA structural element in a viroid-like domain of the hepatitis delta virus. Science. [Internet]. 1989;243:649–52. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2492676. Cited 6 June 2016.
Fu TB, Taylor J. The RNAs of hepatitis delta virus are copied by RNA polymerase II in nuclear homogenates. J Virol. [Internet]. 1993;67:6965–72. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8230419. Cited 6 June 2016.
Lai MMC. RNA replication without RNA-dependent RNA polymerase: surprises from hepatitis delta virus. J Virol. [Internet]. 2005;79:7951–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15956541. Cited 6 June 2016.
Chang J, Nie X, Chang HE, Han Z, Taylor J. Transcription of hepatitis delta virus RNA by RNA polymerase II. J Virol. [Internet]. 2008;82:1118–27. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18032511. Cited 6 July 2016.
Taylor JM. Structure and replication of hepatitis delta virus RNA. Curr Top Microbiol Immunol. [Internet]. 2006;307:1–23. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16903218. Cited 6 June 2016.
Rizzetto M. The adventure of delta. Liver Int. [Internet]. 2016;36(Suppl 1):135–40. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26725911. Cited 6 June 2016.
Taylor JM. Hepatitis delta virus. Virology. [Internet]. 2006;344:71–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16364738. Cited 6 June 2016.
Bonino F, Heermann KH, Rizzetto M, Gerlich WH. Hepatitis delta virus: protein composition of delta antigen and its hepatitis B virus-derived envelope. J Virol. [Internet]. 1986;58:945–50. Available from: http://www.ncbi.nlm.nih.gov/pubmed/3701932. Cited 6 June 2016.
Hughes SA, Wedemeyer H, Harrison PM. Hepatitis delta virus. Lancet (London, England). [Internet]. 2011;378:73–85. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21511329. Cited 6 June 2016.
Wang KS, Choo QL, Weiner AJ, Ou JH, Najarian RC, Thayer RM, et al. Structure, sequence and expression of the hepatitis delta (delta) viral genome. Nature. [Internet]. 1986;323:508–14. Available from: http://www.ncbi.nlm.nih.gov/pubmed/3762705. Cited 6 June 2016.
Kuo MY, Chao M, Taylor J. Initiation of replication of the human hepatitis delta virus genome from cloned DNA: role of delta antigen. J Virol. [Internet]. 1989;63:1945–50. Available from: https://www.ncbi.nlm.nih.gov/pubmed/2649689.
Wang T-C, Chao M. Molecular cloning and expression of the hepatitis delta virus genotype IIb genome. Biochem Biophys Res Commun. [Internet]. 2003;303:357–63. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12646211. Cited 6 June 2016.
Makino S, Chang MF, Shieh CK, Kamahora T, Vannier DM, Govindarajan S, et al. Molecular cloning and sequencing of a human hepatitis delta (delta) virus RNA. Nature. [Internet]. 1987;329:343–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/3627276. Cited 4 July 2016.
Glenn JS, White JM. Trans-dominant inhibition of human hepatitis delta virus genome replication. J Virol. [Internet]. 1991;65:2357–61. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2016764. Cited 4 July 2016.
Sheu SY, Chen KL, Lee YW, Lo SJ. No intermolecular interaction between the large hepatitis delta antigens is required for the secretion with hepatitis B surface antigen: a model of empty HDV particle. Virology. [Internet]. 1996;218:275–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8615035. Cited 4 July 2016.
Sheu G-T. Initiation of hepatitis delta virus (HDV) replication: HDV RNA encoding the large delta antigen cannot replicate. J Gen Virol. [Internet]. 2002;83:2507–13. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12237434. Cited 4 July 2016.
Casey JL. Control of ADAR1 editing of hepatitis delta virus RNAs. Curr Top Microbiol Immunol. [Internet]. 2012;353:123–43. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21732238. Cited 5 July 2016.
Casey JL. RNA editing in hepatitis delta virus. Curr Top Microbiol Immunol. [Internet]. 2006;307:67–89. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16903221. Cited 5 July 2016.
Casey JL, Bergmann KF, Brown TL, Gerin JL. Structural requirements for RNA editing in hepatitis delta virus: evidence for a uridine-to-cytidine editing mechanism. Proc Natl Acad Sci USA. [Internet]. 1992;89:7149–53. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1496009. Cited 5 July 2016.
Casey JL, Gerin JL. Hepatitis D virus RNA editing: specific modification of adenosine in the antigenomic RNA. J Virol. [Internet]. 1995;69:7593–600. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7494266. Cited 5 July 2016.
Chao YC, Chang MF, Gust I, Lai MM. Sequence conservation and divergence of hepatitis delta virus RNA. Virology. [Internet]. 1990;178:384–92. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2219700. Cited 4 July 2016.
Chang MF, Baker SC, Soe LH, Kamahora T, Keck JG, Makino S, et al. Human hepatitis delta antigen is a nuclear phosphoprotein with RNA-binding activity. J Virol. [Internet]. 1988;62:2403–10. Available from: http://www.ncbi.nlm.nih.gov/pubmed/3373572. Cited 5 July 2016.
Mu JJ, Wu HL, Chiang BL, Chang RP, Chen DS, Chen PJ. Characterization of the phosphorylated forms and the phosphorylated residues of hepatitis delta virus delta antigens. J Virol. [Internet]. 1999;73:10540–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10559375. Cited 5 July 2016.
Mu J-J, Tsay Y-G, Juan L-J, Fu T-F, Huang W-H, Chen D-S, et al. The small delta antigen of hepatitis delta virus is an acetylated protein and acetylation of lysine 72 may influence its cellular localization and viral RNA synthesis. Virology. 2004;319:60–70.
Li Y-J, Stallcup MR, Lai MMC. Hepatitis delta virus antigen is methylated at arginine residues, and methylation regulates subcellular localization and RNA replication. J Virol. [Internet]. 2004;78:13325–34. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15542683. Cited 5 July 2016.
Glenn JS, Watson JA, Havel CM, White JM. Identification of a prenylation site in delta virus large antigen. Science. [Internet]. 1992;256:1331–3. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1598578. Cited 5 July 2016.
Huang WH, Chen CW, Wu HL, Chen PJ. Post-translational modification of delta antigen of hepatitis D virus. Curr Top Microbiol Immunol. [Internet]. 2006;307:91–112. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16903222. Cited 5 July 2016.
Huang C-R, Wang RYL, Hsu S-C, Lo SJ. Lysine-71 in the large delta antigen of hepatitis delta virus clade 3 modulates its localization and secretion. Virus Res. 2012;170:75–84.
Hong S-Y, Chen P-J. Phosphorylation of serine 177 of the small hepatitis delta antigen regulates viral antigenomic RNA replication by interacting with the processive RNA polymerase II. J Virol. [Internet]. 2010;84:1430–8. Available from: http://jvi.asm.org/cgi/doi/10.1128/JVI.02083-09. Cited 9 Aug 2017.
Tan K-P, Shih K-N, Lo SJ. Ser-123 of the large antigen of hepatitis delta virus modulates its cellular localization to the nucleolus, SC-35 speckles or the cytoplasm. J Gen Virol [Internet]. 2004;85:1685–94. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15166453. Cited 9 Aug 2017.
Hong S-Y, Chen P-J. Phosphorylation of serine 177 of the small hepatitis delta antigen regulates viral antigenomic RNA replication by interacting with the processive RNA polymerase II. J Virol. [Internet]. 2010;84:1430–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19923176. Cited 5 Jul 2016.
Mu JJ, Chen DS, Chen PJ. The conserved serine 177 in the delta antigen of hepatitis delta virus is one putative phosphorylation site and is required for efficient viral RNA replication. J Virol. [Internet]. 2001;75:9087–95. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11533172. Cited 5 July 2016.
Sharmeen L, Kuo MY, Dinter-Gottlieb G, Taylor J. Antigenomic RNA of human hepatitis delta virus can undergo self-cleavage. J Virol. [Internet]. 1988;62:2674–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2455816. Cited 5 July 2016.
Ferré-D’Amaré AR, Zhou K, Doudna JA. Crystal structure of a hepatitis delta virus ribozyme. Nature. [Internet]. 1998;395:567–74. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9783582. Cited 5 July 2016.
Wu HN, Lai MM. RNA conformational requirements of self-cleavage of hepatitis delta virus RNA. Mol Cell Biol. [Internet]. 1990;10:5575–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2398903. Cited 5 July 2016.
Webb C-HT, Nguyen D, Myszka M, Lupták A, Jimenez RM, Polanco JA, et al. Topological constraints of structural elements in regulation of catalytic activity in HDV-like self-cleaving ribozymes. Sci Rep. [Internet] Nature Publishing Group. 2016;6:28179. Available from: http://www.nature.com/articles/srep28179. Cited 5 July 2016.
Elena SF, Dopazo J, Florest R, Diener TO, Moya A. Phylogeny of viroids, viroidlike satellite RNAs, and the viroidlike domain of hepatitis 6 virus RNA. Evolution (N Y). 1991;88:5631–4.
Tseng C-H, Lai MMC. Hepatitis delta virus RNA replication. Viruses. [Internet]. 2009;1:818–31. Available from: http://www.mdpi.com/1999-4915/1/3/818/. Cited 7 Aug 2017.
Taylor JM, Hepatitis D. Virus replication. Cold spring Harb. Perspect Med. [Internet]. 2015;5:a021568. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26525452. Cited 4 Aug 2017.
Chao M, Wang T-C, Lin C-C, Wang RY-L, Lin W-B, Lee S-E, et al. Analyses of a whole-genome inter-clade recombination map of hepatitis delta virus suggest a host polymerase-driven and viral RNA structure-promoted template-switching mechanism for viral RNA recombination. Oncotarget. [Internet]. 2017; Available from: http://www.oncotarget.com/fulltext/18339. Cited 7 Aug 2017.
Flores R, Grubb D, Elleuch A, Nohales M-Á, Delgado S, Gago S. Rolling-circle replication of viroids, viroid-like satellite RNAs and hepatitis delta virus: variations on a theme. RNA Biol. [Internet]. 2011;8:200–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21358283. Cited 5 July 2016.
Gerlich WH. Medical virology of hepatitis B: how it began and where we are now. Virol J. [Internet]. 2013;10:239. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23870415. Cited 5 July 2016.
Watashi K, Urban S, Li W, Wakita T. NTCP and beyond: opening the door to unveil hepatitis B virus entry. Int J Mol Sci. 2014;15:2892–905.
Li J, Tong S. From DCPD to NTCP: the long journey towards identifying a functional hepatitis B virus receptor. Clin Mol Hepatol. [Internet]. 2015;21:193–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26523264. Cited 5 July 2016.
Li W. The hepatitis B virus receptor. Annu Rev Cell Dev Biol. [Internet]. 2015;31:annurev-cellbio-100814-125241. Available from: http://www.annualreviews.org/doi/10.1146/annurev-cellbio-100814-125241.
Xia YP, Yeh CT, Ou JH, Lai MM. Characterization of nuclear targeting signal of hepatitis delta antigen: nuclear transport as a protein complex. J Virol. [Internet]. 1992;66:914–21. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1731113. Cited 5 July 2016.
Tavanez JP, Cunha C, Silva MCA, David E, Monjardino J, Carmo-Fonseca M. Hepatitis delta virus ribonucleoproteins shuttle between the nucleus and the cytoplasm. RNA. [Internet]. 2002;8:637–46. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12022230. Cited 6 July 2016.
Alves C, Freitas N, Cunha C. Characterization of the nuclear localization signal of the hepatitis delta virus antigen. Virology. 2008;370:12–21.
Yan H, Li W. Sodium taurocholate cotransporting polypeptide acts as a receptor for hepatitis B and D virus. Dig Dis. 2015;33:388–96.
Modahl LE, Macnaughton TB, Zhu N, Johnson DL, Lai MM. RNA-dependent replication and transcription of hepatitis delta virus RNA involve distinct cellular RNA polymerases. Mol Cell Biol. [Internet]. 2000;20:6030–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10913185. Cited 6 July 2016.
Greco-Stewart V, Pelchat M. Interaction of host cellular proteins with components of the Hepatitis Delta virus. Viruses. [Internet]. 2010;2:189–212. Available from: www.mdpi.com/journal/viruses. Cited 6 July 2016.
Wu JC, Chiang TY, Shiue WK, Wang SY, Sheen IJ, Huang YH, et al. Recombination of hepatitis D virus RNA sequences and its implications. Mol Biol Evol. [Internet]. 1999;16:1622–32. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10555293. Cited 7 Aug 2017.
Wang T-C, Chao M. RNA recombination of hepatitis delta virus in natural mixed-genotype infection and transfected cultured cells. J Virol. [Internet]. 2005;79:2221–9. Available from: http://jvi.asm.org/cgi/doi/10.1128/JVI.79.4.2221-2229.2005. Cited 7 Aug 2017.
Lin C-C, Lee C-C, Lin S-H, Huang P-J, Li H-P, Chang Y-S, et al. RNA recombination in hepatitis delta virus: identification of a novel naturally occurring recombinant. J Microbiol Immunol Infect. [Internet]. 2015; Available from: http://linkinghub.elsevier.com/retrieve/pii/S168411821500910X. Cited 7 Aug 2017.
Sy BT, Nguyen HM, Toan NL, Song LH, Tong HV, Wolboldt C, et al. Identification of a natural intergenotypic recombinant hepatitis delta virus genotype 1 and 2 in Vietnamese HBsAg-positive patients. J Viral Hepat. [Internet]. 2015;22:55–63. Available from: http://doi.wiley.com/10.1111/jvh.12228. Cited 7 Aug 2017.
Sureau C, Negro F. The hepatitis delta virus: replication and pathogenesis. J Hepatol. [Internet]. 2016;64:S102–16. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0168827816001203. Cited 7 Aug 2017.
Hwang SB, Lai MM. Isoprenylation mediates direct protein-protein interactions between hepatitis large delta antigen and hepatitis B virus surface antigen. J Virol. [Internet]. 1993;67:7659–62. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8230486. Cited 6 July 2016.
Radjef N, Gordien E, Ivaniushina V, Gault E, Anaïs P, Drugan T, et al. Molecular phylogenetic analyses indicate a wide and ancient radiation of African hepatitis delta virus, suggesting a deltavirus genus of at least seven major clades. J Virol. [Internet]. 2004;78:2537–44. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14963156. Cited 6 July 2016.
Dény P. Hepatitis delta virus genetic variability: from genotypes I, II, III to eight major clades? Curr Top Microbiol Immunol. [Internet]. 2006;307:151–71. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16903225. Cited 6 July 2016.
Le Gal F, Gault E, Ripault M-P, Serpaggi J, Trinchet J-C, Gordien E, et al. Eighth major clade for hepatitis delta virus. Emerg Infect Dis. [Internet]. 2006;12:1447–50. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17073101. Cited 6 July 2016.
Hughes SA, Wedemeyer H, Harrison PM. Hepatitis delta virus. Lancet. 2011;378:73–85.
Farci P. Delta hepatitis: an update. J Hepatol. [Internet]. 2003;39(Suppl 1):S212–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14708706. Cited 6 July 2016.
Rizzetto M, Ponzetto A, Forzani I. Hepatitis delta virus as a global health problem. Vaccine. [Internet]. 1990;8 Suppl:S10-4-3. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2183511. Cited 6 July 2016.
Shakil AO, Hadziyannis S, Hoofnagle JH, Di Bisceglie AM, Gerin JL, Casey JL. Geographic distribution and genetic variability of Hepatitis Delta virus genotype I. Virology. [Internet] Academic Press. 1997;234:160–7. Available from: http://linkinghub.elsevier.com/retrieve/pii/S004268229798644X. Cited 6 July 2016.
Casey JL, Brown TL, Colan EJ, Wignall FS, Gerin JL. A genotype of hepatitis D virus that occurs in northern South America. Proc Natl Acad Sci USA. [Internet]. 1993;90:9016–20. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8415646. Cited 6 July 2016.
Paraná R, Kay A, Molinet F, Viana S, Silva LK, Salcedo JM, et al. HDV genotypes in the western Brazilian Amazon region: a preliminary report. Am J Trop Med Hyg. [Internet]. 2006;75:475–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16968924. Cited 6 July 2016.
Botelho-Souza LF, Souza Vieira D, de Oliveira Dos Santos A, Cunha Pereira AV, Villalobos-Salcedo JM. Characterization of the genotypic profile of Hepatitis Delta virus: isolation of HDV Genotype-1 in the western Amazon region of Brazil. Intervirology. [Internet]. 2015;58:166–71. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26112316. Cited 6 July 2016.
Imazeki F, Omata M, Ohto M. Heterogeneity and evolution rates of delta virus RNA sequences. J Virol. [Internet]. 1990;64:5594–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2214027. Cited 6 July 2016.
Wu JC, Chiang TY, Sheen IJ. Characterization and phylogenetic analysis of a novel hepatitis D virus strain discovered by restriction fragment length polymorphism analysis. J Gen Virol. [Internet]. 1998;79(Pt 5):1105–13. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9603325. Cited 6 July 2016.
Ivaniushina V, Radjef N, Alexeeva M, Gault E, Semenov S, Salhi M, et al. Hepatitis delta virus genotypes I and II cocirculate in an endemic area of Yakutia, Russia. J Gen Virol. [Internet]. 2001;82:2709–18. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11602783. Cited 6 July 2016.
Alvarado-Mora MV, Fernandez MFG, Gomes-Gouvêa MS, de Azevedo Neto RS, Carrilho FJ, Pinho JRR. Hepatitis B (HBV), hepatitis C (HCV) and hepatitis delta (HDV) viruses in the Colombian population--how is the epidemiological situation? PLoS One. [Internet]. 2011;6:e18888. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21559488. Cited 6 July 2016.
Gomes-Gouvêa MS, do Pereira Soares MC, Guedes de Carvalho Mello IMV, Brito EMF, de Pereira Moia LJM, Bensabath G, et al. Hepatitis D and B virus genotypes in chronically infected patients from the eastern Amazon Basin. Acta Trop. [Internet]. 2008;106:149–55. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18420172. Cited 6 July 2016.
Gomes-Gouvêa MS, Soares MCP, Bensabath G, de Carvalho-Mello IMVG, Brito EMF, Souza OSC, et al. Hepatitis B virus and hepatitis delta virus genotypes in outbreaks of fulminant hepatitis (Labrea black fever) in the western Brazilian Amazon region. J Gen Virol. [Internet]. 2009;90:2638–43. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19605587. Cited 6 July 2016.
Alvarado-Mora MV, Romano CM, Gomes-Gouvêa MS, Gutierrez MF, Carrilho FJ, Pinho JRR. Dynamics of hepatitis D (delta) virus genotype 3 in the Amazon region of South America. Infect Genet Evol. [Internet]. 2011;11:1462–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21645647. Cited 6 July 2016.
Ma S-P, Sakugawa H, Makino Y, Tadano M, Kinjo F, Saito A. The complete genomic sequence of hepatitis delta virus genotype IIb prevalent in Okinawa, Japan. J Gen Virol. [Internet]. 2003;84:461–4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12560580. Cited 6 July 2016.
Barros LMF, Gomes-Gouvêa MS, Pinho JRR, Alvarado-Mora MV, Dos Santos A, Mendes-Corrêa MCJ, et al. Hepatitis Delta virus genotype 8 infection in Northeast Brazil: inheritance from African slaves? Virus Res. [Internet]. 2011;160:333–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21798297. Cited 6 July 2016.
Viana S, Paraná R, Moreira RC, Compri AP, Macedo V. High prevalence of hepatitis B virus and hepatitis D virus in the western Brazilian Amazon. Am J Trop Med Hyg. [Internet]. 2005;73:808–14. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16222030. Cited 6 July 2016.
Crispim MAE, Fraiji NA, Campello SC, Schriefer NA, Stefani MMA, Kiesslich D. Molecular epidemiology of hepatitis B and hepatitis delta viruses circulating in the western Amazon region, North Brazil. BMC Infect Dis. [Internet]. 2014;14:94. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24555665. Cited 6 July 2016.
Fonseca JC, Simonetti SR, Schatzmayr HG, Castejón MJ, Cesário AL, Simonetti JP. Prevalence of infection with hepatitis delta virus (HDV) among carriers of hepatitis B surface antigen in Amazonas state, Brazil. Trans R Soc Trop Med Hyg. [Internet]. 1988;82:469–71. Available from: http://www.ncbi.nlm.nih.gov/pubmed/3232188. Cited 6 July 2016.
Vasconcelos M, Pereira DB, Paraná R, Villalobos-Salcedo JM. Clinic and laboratory analysis of patients with hepatitis delta in Amazon region, Brazil. J Med Med Sci. [Internet]. 2012;3:263–9. Available from: http://www.interesjournals.org/JMMS. Cited 6 July 2016.
Braga WSM. Hepatitis B and D virus infection within Amerindians ethnic groups in the Brazilian Amazon: epidemiological aspects. Rev Soc Bras Med Trop. [Internet]. 2004;37(Suppl 2):9–13. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15586891. Cited 6 July 2016.
Braga WSM, da Castilho MC, Borges FG, Leão JRDT, de Martinho ACS, Rodrigues IS, et al. Hepatitis D virus infection in the Western Brazilian Amazon - far from a vanishing disease. Rev Soc Bras Med Trop. [Internet]. 2012;45:691–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23295870. Cited 6 July 2016.
Mendes-Correa MC, Gomes-Gouvêa MS, Alvarado-Mora MV, Da Silva MH, Lázari C, Cavalcanti NCS, et al. Hepatitis delta in HIV/HBV co-infected patients in Brazil: is it important? Int J Infect Dis. 2011;15:e828–32.
Farci P, Niro GA. Clinical features of hepatitis D. Semin Liver Dis. [Internet]. 2012;32:228–36. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22932971. Cited 7 July 2016.
Smedile A, Verme G, Cargnel A, Dentico P, Opolon P, Vergani D, et al. INFLUENCE OF DELTA INFECTION ON SEVERITY OF HEPATITIS B. Lancet. [Internet] Elsevier. 1982;320:945–7. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0140673682901568. Cited 7 July 2016.
Rizzetto M, Durazzo M. Hepatitis delta virus (HDV) infections. Epidemiological and clinical heterogeneity. J Hepatol. [Internet]. 1991;13(Suppl 4):S116–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1822503. Cited 7 July 2016.
Romeo R, Del Ninno E, Rumi M, Russo A, Sangiovanni A, de Franchis R, et al. A 28-year study of the course of hepatitis Δ infection: a risk factor for cirrhosis and Hepatocellular carcinoma. Gastroenterology. 2009;136:1629–38.
Grabowski J, Wedemeyer H. Hepatitis delta: immunopathogenesis and clinical challenges. Dig Dis. [Internet]. 2010;28:133–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20460901. Cited 7 July 2016.
Smedile A, Rizzetto M, Gerin JL. Advances in hepatitis D virus biology and disease. Prog Liver Dis. [Internet]. 1994;12:157–75. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7746872. Cited 7 July 2016.
Pascarella S, Negro F. Hepatitis D virus: An update. Liver Int. 2011;31:7–21.
Flodgren E, Bengtsson S, Knutsson M, Strebkova EA, Kidd AH, Alexeyev OA, et al. Recent high incidence of fulminant hepatitis in Samara, Russia: molecular analysis of prevailing hepatitis B and D virus strains. J Clin Microbiol. [Internet]. 2000;38:3311–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10970376. Cited 7 July 2016.
Casey JL, Niro GA, Engle RE, Vega A, Gomez H, McCarthy M, et al. Hepatitis B virus (HBV)/hepatitis D virus (HDV) coinfection in outbreaks of acute hepatitis in the Peruvian Amazon basin: the roles of HDV genotype III and HBV genotype F. J Infect Dis. [Internet]. 1996;174:920–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8896491. Cited 7 July 2016.
Schaper M, Rodriguez-Frias F, Jardi R, Tabernero D, Homs M, Ruiz G, et al. Quantitative longitudinal evaluations of hepatitis delta virus RNA and hepatitis B virus DNA shows a dynamic, complex replicative profile in chronic hepatitis B and D. J Hepatol. 2010;52:658–64.
Lozano JL, Crespo J, de la Cruz F, Casafont F, Lopez-Arias MJ, Martín-Ramos L, et al. Correlation between hepatitis B viremia and the clinical and histological activity of chronic delta hepatitis. Med Microbiol Immunol. [Internet]. 1994;183:159–67. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7997189. Cited 7 July 2016.
Fattovich G, Giustina G, Christensen E, Pantalena M, Zagni I, Realdi G, et al. Influence of hepatitis delta virus infection on morbidity and mortality in compensated cirrhosis type B. The European concerted action on viral hepatitis (Eurohep). Gut. [Internet]. 2000;46:420–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10673308. Cited 7 July 2016.
Sánchez-Tapias JM, Mas A, Costa J, Bruguera M, Mayor A, Ballesta AM, et al. Recombinant alpha 2c-interferon therapy in fulminant viral hepatitis. J Hepatol. [Internet]. 1987;5:205–10. Available from: http://www.ncbi.nlm.nih.gov/pubmed/3693864. Cited 7 Jul 2016.
Buti M, Esteban R, Roggendorf M, Fernandez J, Jardi R, Rashofer R, et al. Hepatitis D virus RNA in acute delta infection: serological profile and correlation with other markers of hepatitis D virus infection. Hepatology. [Internet]. 1988;8:1125–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/3417234. Cited 7 July 2016.
Jardi R, Buti M, Rodriguez-Frias F, Garcia-Lafuente A, Sjogren MH, Esteban R, et al. Clinical significance of two forms of IgM antibody to hepatitis delta virus. Hepatology. [Internet]. 1991;14:25–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2066073. Cited 7 July 2016.
Buti M, Lopez-Talavera JC, Allende H, Jardi R, Esteban R, Guardia J. Serological diagnosis of chronic delta infection: correlation between serological markers and hepatitis delta virus RNA in hepatic tissue. Prog Clin Biol Res. [Internet]. 1993;382:319–23. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8502697. Cited 7 July 2016.
Jardi R, Buti M, Rodriguez F, Cotrina M, Allende H, Esteban R, et al. Comparative analysis of serological markers of chronic delta infection: HDV-RNA, serum HDAg and anti-HD IgM. J Virol Methods. [Internet]. 1994;50:59–66. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7714059. Cited 7 July 2016.
Jardi R, Buti M, Cotrina M, Rodriguez F, Allende H, Esteban R, et al. Determination of hepatitis delta virus RNA by polymerase chain reaction in acute and chronic delta infection. Hepatology. [Internet] WB Saunders. 1995;21:25–9. Available from: http://doi.wiley.com/10.1002/hep.1840210106. Cited 7 July 2016.
Buti M, Jardi R, Allende H, Cotrina M, Rodriguez F, Viladomiu L, et al. Chronic delta hepatitis: is the prognosis worse when associated with hepatitis C virus and human immunodeficiency virus infections? J Med Virol. [Internet]. 1996;49:66–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8732862. Cited 7 July 2016.
Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. (London, England) [Internet] NIH Public Access. 2008;371:838–51. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18328931. Cited 8 July 2016.
Niro GA, Smedile A. Current concept in the pathophysiology of hepatitis delta infection. Curr Infect Dis Rep. [Internet]. 2012;14:9–14. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22161240. Cited 8 July 2016.
Verme G, Amoroso P, Lettieri G, Pierri P, David E, Sessa F, et al. A histological study of hepatitis delta virus liver disease. Hepatology. [Internet]. 1986;6:1303–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/3793008. Cited 8 July 2016.
Heidrich B, Yurdaydın C, Kabaçam G, Ratsch BA, Zachou K, Bremer B, et al. Late HDV RNA relapse after peginterferon alpha-based therapy of chronic hepatitis delta. Hepatology. [Internet]. 2014;60:87–97. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24585488. Cited 8 July 2016.
Rizzetto M, Alavian SM. Hepatitis delta: the rediscovery. Clin Liver Dis. 2013;17:475–87.
Yurdaydin C. Treatment of chronic delta hepatitis. Semin Liver Dis. [Internet]. 2012;32:237–44. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22932972. Cited 8 July 2016.
Heller T, Rotman Y, Koh C, Clark S, Haynes-Williams V, Chang R, et al. Long-term therapy of chronic delta hepatitis with peginterferon alfa. Aliment Pharmacol Ther. [Internet]. 2014;40:93–104. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24815494. Cited 8 July 2016.
Niro GA, Ciancio A, Tillman HL, Lagget M, Olivero A, Perri F, et al. Lamivudine therapy in chronic delta hepatitis: a multicentre randomized-controlled pilot study. Aliment Pharmacol Ther. [Internet]. 2005;22:227–32. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16091060. Cited 8 July 2016.
Erhardt A, Gerlich W, Starke C, Wend U, Donner A, Sagir A, et al. Treatment of chronic hepatitis delta with pegylated interferon-α2b. Liver Int. [Internet] Blackwell Publishing Ltd. 2006;26:805–10. Available from: http://doi.wiley.com/10.1111/j.1478-3231.2006.01279.x. Cited 8 July 2016.
Lau DT, Doo E, Park Y, Kleiner DE, Schmid P, Kuhns MC, et al. Lamivudine for chronic delta hepatitis. Hepatology. [Internet]. W.B. Saunders. 1999;30:546–9. Available from: http://doi.wiley.com/10.1002/hep.510300217. Cited 8 July 2016.
Kabaçam G, Onder FO, Yakut M, Seven G, Karatayli SC, Karatayli E, et al. Entecavir treatment of chronic hepatitis D. Clin Infect Dis. [Internet]. 2012;55:645–50. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22573857. Cited 8 July 2016.
Gunsar F, Akarca US, Ersoz G, Kobak AC, Karasu Z, Yuce G, et al. Two-year interferon therapy with or without ribavirin in chronic delta hepatitis. Antivir Ther. [Internet]. 2005;10:721–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16218171. Cited 8 July 2016.
Yurdaydin C, Bozkaya H, Karaaslan H, Onder FO, Erkan OE, Yalçin K, et al. A pilot study of 2 years of interferon treatment in patients with chronic delta hepatitis. J Viral Hepat. [Internet]. 2007;14:812–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17927618. Cited 8 Jul 2016.
Samiullah S, Bikharam D, Nasreen. Treatment of chronic hepatitis delta virus with peg-interferon and factors that predict sustained viral response. World J Gastroenterol. [Internet] Baishideng Publishing Group Inc. 2012;18:5793–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23155322. Cited 8 July 2016.
Gheorghe L, Iacob S, Simionov I, Vadan R, Constantinescu I, Caruntu F, et al. Weight-based dosing regimen of peg-interferon α-2b for chronic hepatitis delta: a multicenter Romanian trial. J Gastrointestin Liver Dis. [Internet]. 2011;20:377–82. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22187703. Cited 8 July 2016.
Koh C, Canini L, Dahari H, Zhao X, Uprichard SL, Haynes-Williams V, et al. Oral prenylation inhibition with lonafarnib in chronic hepatitis D infection: a proof-of-concept randomised, double-blind, placebo-controlled phase 2A trial. Lancet Infect Dis. [Internet]. 2015;15:1167–74. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1473309915000742. Cited 26 July 2017.
Rizzetto M, Niro GA. Myrcludex B, a novel therapy for chronic hepatitis D? J Hepatol. [Internet]. 2016;65:465–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27538763. Cited 26 July 2017.
Bogomolov P, Alexandrov A, Voronkova N, Macievich M, Kokina K, Petrachenkova M, et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: first results of a phase Ib/IIa study. J Hepatol. [Internet]. 2016;65:490–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27132170. Cited 26 July 2017.
Abeywickrama-Samarakoon N, Cortay J-C, Dény P. The hepatitis D satellite virus of hepatitis B virus: half-opening a new era to control viral infection? Curr Opin Infect Dis. [Internet]. 2016;29:645–53. Available from: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00001432-201612000-00014. Cited 26 July 2017.
Rizzetto M, Ciancio A. The prenylation inhibitor, lonafarnib: a new therapeutic strategy against hepatitis delta. Lancet Infect Dis. [Internet]. 2015;15:1119–20. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1473309915001553. Cited 26 July 2017.
Giersch K, Homs M, Volz T, Helbig M, Allweiss L, Lohse AW, et al. Both interferon alpha and lambda can reduce all intrahepatic HDV infection markers in HBV/HDV infected humanized mice. Sci Rep. [Internet]. 2017;7:3757. Available from: http://www.nature.com/articles/s41598-017-03946-9. Cited 26 July 2017.
Borzacov LMP, de Figueiredo Nicolete LD, Souza LFB, dos Santos AO, Vieira DS, Salcedo JMV. Treatment of hepatitis delta virus genotype 3 infection with peg-interferon and entecavir. Int J Infect Dis. [Internet]. 2016;46:82–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27005283. Cited 7 Aug 2017.
Ciancio A, Rizzetto M. Chronic hepatitis D at a standstill: where do we go from here? Nat Rev Gastroenterol Hepatol. [Internet]. 2014;11:68–71. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24019153. Cited 8 July 2016.
Manesis EK, Vourli G, Dalekos G, Vasiliadis T, Manolaki N, Hounta A, et al. Prevalence and clinical course of hepatitis delta infection in Greece: a 13-year prospective study. J Hepatol. 2013;59:949–56.
Abbas Z, Qureshi M, Hamid S, Jafri W. Hepatocellular carcinoma in hepatitis D: does it differ from hepatitis B monoinfection? Saudi J Gastroenterol. [Internet]. 2012;18:18–22. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22249087. Cited 8 July 2016.
Heidrich B, Manns MP, Wedemeyer H. Treatment options for hepatitis delta virus infection. Curr Infect Dis Rep. [Internet]. 2013;15:31–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23242761. Cited 8 July 2016.
Ji J, Sundquist K, Sundquist J. A population-based study of hepatitis D virus as potential risk factor for hepatocellular carcinoma. J Natl Cancer Inst. [Internet]. 2012;104:790–2. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22423008. Cited 8 July 2016.
Freitas N, Salisse J, Cunha C, Toshkov I, Menne S, Gudima SO. Hepatitis delta virus infects the cells of hepadnavirus-induced hepatocellular carcinoma in woodchucks. Hepatology. [Internet]. 2012;56:76–85. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22334419. Cited 8 July 2016.
Cross TJS, Rizzi P, Horner M, Jolly A, Hussain MJ, Smith HM, et al. The increasing prevalence of hepatitis delta virus (HDV) infection in South London. J Med Virol. [Internet]. 2008;80:277–82. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18098143. Cited 8 July 2016.
Zachou K, Yurdaydin C, Drebber U, Dalekos GN, Erhardt A, Cakaloglu Y, et al. Quantitative HBsAg and HDV-RNA levels in chronic delta hepatitis. Liver Int. [Internet]. 2010;30:430–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19840253. Cited 8 July 2016.
Wranke A, Heidrich B, Ernst S, Calle Serrano B, Caruntu FA, Curescu MG, et al. Anti-HDV IgM as a marker of disease activity in hepatitis delta. PLoS One. [Internet]. 2014;9:e101002. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25072849. Cited 8 July 2016.
Farci P, Roskams T, Chessa L, Peddis G, Mazzoleni AP, Scioscia R, et al. Long-term benefit of interferon α therapy of chronic hepatitis D: regression of advanced hepatic fibrosis. Gastroenterology. 2004;126:1740–9.
Madejón A, Cotonat T, Bartolomé J, Castillo I, Carreño V. Treatment of chronic hepatitis D virus infection with low and high doses of interferon-alpha 2a: utility of polymerase chain reaction in monitoring antiviral response. Hepatology. [Internet]. 1994;19:1331–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8188163. Cited 8 July 2016.
Garripoli A, Di Marco V, Cozzolongo R, Costa C, Smedile A, Fabiano A, et al. Ribavirin treatment for chronic hepatitis D: a pilot study. Liver. [Internet]. 1994;14:154–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8078394. Cited 8 July 2016.
Yurdaydin C, Bozkaya H, Gürel S, Tillmann HL, Aslan N, Okçu-Heper A, et al. Famciclovir treatment of chronic delta hepatitis. J Hepatol. 2002;37:266–71.
Castelnau C, Le Gal F, Ripault M-P, Gordien E, Martinot-Peignoux M, Boyer N, et al. Efficacy of peginterferon alpha-2b in chronic hepatitis delta: relevance of quantitative RT-PCR for follow-up. Hepatology. [Internet]. 2006;44:728–35. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16941695. Cited 8 July 2016.
Niro GA, Ciancio A, Gaeta GB, Smedile A, Marrone A, Olivero A, et al. Pegylated interferon alpha-2b as monotherapy or in combination with ribavirin in chronic hepatitis delta. Hepatology. [Internet]. 2006;44:713–20. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16941685. Cited 8 July 2016.
Canbakan B, Senturk H, Tabak F, Akdogan M, Tahan V, Mert A, et al. Efficacy of interferon alpha-2b and lamivudine combination treatment in comparison to interferon alpha-2b alone in chronic delta hepatitis: a randomized trial. J Gastroenterol Hepatol. [Internet]. 2006;21:657–63. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16677149. Cited 8 July 2016.
Yurdaydin C, Bozkaya H, Onder FO, Sentürk H, Karaaslan H, Akdoğan M, et al. Treatment of chronic delta hepatitis with lamivudine vs lamivudine + interferon vs interferon. J Viral Hepat. [Internet]. 2008;15:314–21. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18307594. Cited 8 July 2016.
Ormeci N, Bölükbaş F, Erden E, Coban S, Ekiz F, Erdem H, et al. Pegylated interferon alfa-2B for chronic delta hepatitis: 12 versus 24 months. Hepatogastroenterology. [Internet]. 2011;58:1648–53. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22086695. Cited 8 July 2016.
Wedemeyer H, Yurdaydìn C, Dalekos GN, Erhardt A, Çakaloğlu Y, Değertekin H, et al. Peginterferon plus adefovir versus either drug alone for hepatitis delta. N Engl J Med. [Internet]. 2011;364:322–31. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21268724. Cited 8 July 2016.
Karaca C, Soyer OM, Baran B, Ormeci AC, Gokturk S, Aydin E, et al. Efficacy of pegylated interferon-α treatment for 24 months in chronic delta hepatitis and predictors of response. Antivir Ther. [Internet]. 2013;18:561–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22976528. Cited 8 July 2016.
Acknowledgements
FAPERO and FIOCRUZ Rondônia for supporting the submission decision of the article.
Funding
Not aplicable.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Search strategy and selection criteria
The references for this review were identified by searching PubMed for articles published from January 1977 to June 2016, the use of the terms “Virus Hepatitis Delta”, “delta antigen” and “HDV”. Articles resulting from these surveys and relevant references cited in these articles were reviewed. Articles published in English and Portuguese were included.
Author information
Authors and Affiliations
Contributions
Doctors JMVS and DSV were responsible for the general guidance and coordination of work. LFSB was responsible for research references and elaboration of the manuscript. MPAV researched references and elaboration of manuscript. AOdS participated in elaboration of manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable in this section.
Consent for publication
Not applicable in this section.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Botelho-Souza, L.F., Vasconcelos, M.P.A., dos Santos, A.d. et al. Hepatitis delta: virological and clinical aspects. Virol J 14, 177 (2017). https://doi.org/10.1186/s12985-017-0845-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-017-0845-y