
Abstract
Background
Over the past decades, positron emission tomography (PET) imaging has become an increasingly useful research modality in the field of multiple sclerosis (MS) research, as PET can visualise molecular processes, such as neuroinflammation, in vivo. The second generation PET radioligand [18F]DPA714 binds with high affinity to the 18-kDa translocator-protein (TSPO), which is mainly expressed on activated microglia. The aim of this proof of concept study was to evaluate this in vivo marker of neuroinflammation in primary and secondary progressive MS.
Methods
All subjects were genotyped for the rs6971 polymorphism within the TSPO gene, and low-affinity binders were excluded from participation in this study. Eight patients with progressive MS and seven age and genetic binding status matched healthy controls underwent a 60 min dynamic PET scan using [18F]DPA714, including both continuous on-line and manual arterial blood sampling to obtain metabolite-corrected arterial plasma input functions.
Results
The optimal model for quantification of [18F]DPA714 kinetics was a reversible two-tissue compartment model with additional blood volume parameter. For genetic high-affinity binders, a clear increase in binding potential was observed in patients with MS compared with age-matched controls. For both high and medium affinity binders, a further increase in binding potential was observed in T2 white matter lesions compared with non-lesional white matter. Volume of distribution, however, did not differentiate patients from healthy controls, as the large non-displaceable compartment of [18F]DPA714 masks its relatively small specific signal.
Conclusion
The TSPO radioligand [18F]DPA714 can reliably identify increased focal and diffuse neuroinflammation in progressive MS when using plasma input-derived binding potential, but observed differences were predominantly visible in high-affinity binders.
Similar content being viewed by others

Background
Conventional magnetic resonance imaging [1] has a central role in the diagnosis of multiple sclerosis (MS), based on its high sensitivity for identifying MS-specific demyelinating lesions in the brain and spinal cord [2]. In patients with a relapsing-remitting disease course, radiological monitoring is based on the development of new MS lesions on T2-weighted MRI and the presence of gadolinium-enhancing lesions on T1-weighted MRI, demonstrating disruption of the blood-brain barrier which is indicative of active neuroinflammation [3]. However, these focal lesions as seen on MRI cannot fully explain the neurological and cognitive deficits in patients with MS [4]. In addition, in both primary and secondary progressive MS, neuroinflammation usually is not characterised by focal active demyelinating lesions [5]. Animal model and human post-mortem studies have identified a more diffuse and low-grade neuroinflammation behind an intact blood-brain barrier, not depicted by conventional MRI [6, 7]. Consequently, there is a need for novel imaging techniques.
Over the past decades, positron emission tomography (PET) imaging has become an increasingly useful research modality in the field of MS research, as PET can visualise molecular processes, such as neuroinflammation, in vivo [8, 9]. Specifically, the 18-kDa translocator-protein (TSPO) receptor, which is present on the mitochondrial membrane of microglial cells, with additional binding sites in monocytes, astrocytes and vascular endothelium, has shown potential as a target for imaging neuroinflammation with PET [10]. Using the first-generation TSPO PET tracer (R)-[11C]PK11195, increased TSPO expression has been demonstrated in both MS lesions and normal appearing brain tissue of patients with MS, which was associated with increased disability and disease progression in all phases of the disease [11,12,13,14,15,16]. Unfortunately, (R)-[11C]PK11195 suffers from poor brain penetration and high nonspecific binding in the brain, hindering accurate quantification [12]. Over the years, a large number of second-generation TSPO tracers have been developed, such as [11C]PBR28, [18F]PBR111, [11C]DPA713, [18F]GE180 and [18F]FEDAA1106. These novel tracers have an improved signal-to-noise ratio, resulting from lower lipophilicity and therefore less non-specific binding to e.g. plasma proteins and improved blood-brain barrier passage [17,18,19,20,21]. It has been shown that the second-generation radioligand [18F]DPA714 provides increased bioavailability to brain tissue and indeed has a higher signal-to-noise ratio [22, 23]. One limitation of second-generation tracers, including [18F]DPA714, is their genetically determined binding affinity, resulting from a single nuclide polymorphism rs6971 within the TSPO gene. Approximately 50% of the general population is a genetically determined high-affinity binder (HAB), 30–40% a medium-affinity binder (MAB) and 10–20% a low-affinity binder (LAB) [24].
To date, quantification of cerebral [18F]DPA714 binding has been described in healthy controls [25] and in patients with Alzheimer’s disease [26], with both studies agreeing on a reversible two-tissue compartment model with blood volume parameter (2T4k_VB) as the optimal kinetic model. In addition, pseudo-quantitative analyses have been performed in post-stroke [27] and Alzheimer’s disease patients [28]. Moreover, the simplified reference tissue model, using cerebellar grey matter as a reference region, has been found to reliably assess binding potential in Alzheimer’s disease [26]. However, the pathophysiology of MS violates the assumptions underlying reference tissue models: firstly, no single brain region can be used as a reference region in MS, as no region can be assumed to be devoid of specific TSPO binding, and secondly, the blood volume contribution is not negligible throughout the MS brain due to potential disruption of the blood-brain barrier [29]. The latter will also influence the kinetics of the tracer making reference tissue models unreliable [30].
The aim of this proof of concept study was to identify the optimal plasma input tracer kinetic model for characterising in vivo [18F]DPA714 kinetics in patients with primary and secondary progressive MS and to evaluate whether the kinetic parameters estimated from this model can be used to quantify TSPO binding in this patient group.
Methods
Participants
Patients diagnosed with primary or secondary progressive MS, according to the 2010 revisions of the McDonald criteria, and healthy controls were recruited from the MS Center, VU University Medical Center, Amsterdam, between January 2015 and February 2016. All subjects were genotyped for the rs6971 polymorphism within the TSPO gene, which predicts binding affinity to the second generation TSPO tracers [24]. Only genetically high and medium affinity binders (HAB and MAB) were included, and genetically low-affinity binders (LAB) were excluded. Further exclusion criteria for all subjects were a medical history of relevant neurological or auto-immune disease (other than MS); known cardiac, haematological, oncological or renal diseases; current or previous alcohol or drug abuse; recent treatment with immunomodulating drugs; or the use of benzodiazepines. None of the patients with progressive MS were on disease-modifying therapy, and no patients were treated with intravenous methylprednisolone for the last 3 months prior to the PET scans. All subjects had normal physical examination and all healthy controls a normal neurological examination.
This study was approved by the Medical Ethics Review Board of the VU University Medical Center and was registered under number 2014.356. All subjects gave written informed consent prior to participation.
MRI acquisition
MR imaging was performed on a 3 Tesla Ingenuity TF PET-MR system (Philips Medical Systems, Best, The Netherlands) on the same day as the PET-CT acquisition. MRI analysis included 3D T1 (repetition time 7.9 ms, echo time 4.5 ms, flip angle 8°, measured voxel size 1 × 1 × 1 mm3) for region of interest (ROI) definition and 3D T2 Fluid Attenuated Inversion Recovery (FLAIR) (repetition time 4800 ms, echo time 279 ms, inversion time 1650 ms, measured voxel size 0.9 × 0.9 × 1.1 mm3) for lesion segmentation. In patients, additional post-contrast 2D T1 sequences (repetition time 600 ms, echo time 10 ms, measured voxel size 0.56 × 0.44 × 5 mm3) were acquired.
PET acquisition and reconstruction
Radiosynthesis of [18F]DPA714 was performed with an in-house built automatic apparatus using procedures described previously [26]. PET scans were acquired on an Ingenuity TF PET-CT scanner (Philips Medical Systems, Best, The Netherlands). Following a low-dose CT scan for attenuation correction, a bolus of 263 ± 13 MBq [18F]DPA714, with a mean molar activity of 39 ± 20 GBq/μmol, was injected intravenously at 0.8 mL/sec with an automated infusion pump. Simultaneously, a 60-min PET dynamic emission scan was started. The emission scan was collected in list mode and rebinned into 19 frames (1 × 15, 3 × 5, 3 × 10, 4 × 60, 2 × 150, 2 × 300, 4 × 600 s). PET data were reconstructed to a final voxel size of 2 × 2 × 2 mm3 using standard scanner software (BLOB-OS-TF), which incorporates standard corrections for attenuation, scatter and randoms.
Arterial blood was withdrawn continuously from the radial artery during the entire scan (using an automated pump for the first 5 min at 5 mL/min and from 5 to 60 min at 2.5 mL/min), and in addition, manual samples were collected at six time points (5, 10, 20, 30, 40 and 60 min) [31]. Continuous whole blood time activity curves (TACs) were corrected for plasma to whole blood ratios, metabolites and time delay to obtain metabolite-corrected arterial plasma input functions.
Data analysis
MS white matter lesions were automatically segmented on the FLAIR and T1-weighted images using kNN-TTP [32]. Lesion filling of the T1-weighted image was obtained using LEAP [33], after which the filled T1-weighted image was registered to the PET scan using the VINCI software package [34, 35]. Grey matter, white matter and cerebral spinal fluid segmentation was performed automatically using SPM8 implemented in PVElab [36]. ROIs were defined according to the Hammers template on the co-registered 3D T1 MRI [37]. Regional TACs were extracted by superimposing the MR-derived ROIs onto the dynamic PET images. Volume weighted larger ROIs were also defined to assess the impact of ROI size on quantitative analyses. The lesion masks were subtracted from segmented white matter to define non-lesional white matter.
Kinetic modelling was performed using standard single-tissue (1T2k) together with reversible and irreversible two-tissue (2T3k and 2T4k) compartment models, both with and without blood volume parameter (VB) [38]. It has been hypothesised that some binding to the vascular endothelium exists for TSPO tracers, which can be characterised by slow irreversible binding (Kb) [39, 40]. Therefore, a 2T4k_VB with an irreversible vascular binding component (1T1k) was also tested. Akaike information criterion (AIC) was used to compare model fits and to identify the optimal tracer kinetic model. Reliability of parameter estimates was determined by the percentage standard deviation (%SD) of these estimates, with a cut-off of 25% for the generally robust K1, and 50% for the other parameters. Estimates for K1, BPND (k3/k4), VT and Kb from the optimal model were used to assess group differences between patients and controls, and between genetically high and medium affinity binders (HAB and MAB).
Statistical analysis
Group differences were evaluated using the Mann-Whitney U test in SPSS 22.0 (IBM Corp., Armonk, NY). Due to the explorative nature of this proof of concept study, we did not correct for multiple comparisons. To evaluate regional differences within the patient group, a Wilcoxon signed-rank test was used.
Results
Demographics
At screening, all subjects were genotyped for the rs6971 polymorphism within the TSPO gene and low-affinity binders were excluded from participation in this study. In total, eight patients diagnosed with primary or secondary progressive MS and seven healthy controls completed the whole study protocol. Demographic information is reported in Table 1. Patients included were four HAB and four MAB, with a mean age of 53 ± 3 years and a wide variation in T2 lesion load, ranging from a few solitary lesions to widespread confluent lesions (3.6 to 60.6 cm2). In only two patients, one small enhancing lesion could be identified, not uncommon in primary or secondary progressive MS. In healthy controls, genetic binding status was similarly distributed: three HAB and four MAB. The mean age for healthy controls was 52 ± 4 years, which was not significantly different from the patients with MS (p = 0.418).
Tracer kinetics
Based on the AIC, for the majority of subjects, the preferred model for quantification of [18F]DPA714 was 2T4k_VB_1T1k, see Fig. 1. However, when evaluating reliability of the parameter estimates, for a large number of regions, K1, BPND and/or Kb could not be reliably estimated, with either a high %SD or values touching the boundaries (see Additional files 1 and 2). This was even the case for subjects in which 2T4k_VB_1T1k was the preferred model according to AIC, and it was true not only for small, but also for large ROIs. As model fits for 2T4k_VB_1T1k and 2T4k_VB were comparable (see Fig. 2) and 2T4k_VB did result in reliable parameter estimates, the latter model was used for further analyses.

Model preference. Number of subjects per preferred model according to the Akaike information criterion. All subjects preferred a reversible two-tissue compartment model (2T4k), primarily for the model including blood volume parameter [38] and a vascular binding component (1T1k)

Model fit comparisons. Representative model fits for the frontal cortex of a medium affinity binding healthy control, the thalamic grey matter of a high-affinity binding MS patient and the T2 lesions of a medium-affinity binding MS patient. The open circles represent the measured time-activity curves (TAC) and the solid grey and dashed black lines represent the fits for the reversible two-tissue compartment model without and with vascular binding component (2T4k_VB and 2T4k_VB_1T1k)
Volume of distribution
Mean regional VT values were on average 1.6 times higher in HABs than in MABs (Fig. 3a). There was no clear regional difference in VT between patients and healthy controls for both HABs and MABs, as Fig. 3a illustrates for the MS white matter lesions, non-lesional white matter and whole brain grey matter. Moreover, very limited within-subject variation in VT was observed, even for the patients with MS. Figure 3b illustrates that all subjects showed a similar distribution pattern for VT throughout the brain. In addition, a strong correlation between VT and K1 was seen (Fig. 4a), whereas no correlations were found between VT and both k2 and k3/k4.

Volume of distribution. a Volume of distribution (VT) for total grey matter non-lesional white matter (WM) and T2 defined white matter lesions for the reversible two-tissue compartment model (2T4k_VB). High-affinity binding (HAB) subjects in red have a 1.5 to 2-fold higher VT than medium affinity binding (MAB) subjects in blue. No difference in VT was seen between multiple sclerosis patients (MS; closed symbols) and healthy controls (HC; open symbols) for both HABs and MABs. b VT across various regions of all patients and controls. The intra-subject variation appears to be much smaller than the inter-subject variation. c The reversible two-tissue compartment model with vascular binding component (2T4k_VB_1T1k) also identified an increase in VT for HAB subjects compared with MAB subjects, but no difference in mean regional VT for patients compared to controls for either HABs or MABs

Correlation for VT and K1. For both the reversible two-tissue compartment model a without and b with vascular binding component (2T4k_VB and 2T4k_VB_1T1k), there was a strong correlation between VT and K1 in both grey and white matter, independent of binding status
Finally, regional VT values for the 2T4k_VB_1T1k model were evaluated (Fig. 3c). As expected, the added vascular binding component decreased the VT. As the regional VT values for the two models were strongly correlated for the large ROIs, there was also no difference in 2T4k_VB_1T1k VT between patients and healthy controls for both the HABs and MABs (Fig. 3c). It is of interest to know that also the correlation between VT and K1 as seen for the simpler model remained the same for this model incorporating a vascular binding component (Fig. 4b).
Binding potential
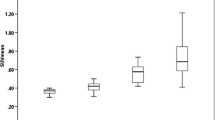
BPND (k3/k4) estimates for the larger ROIs were reliable using the 2T4k_VB model, as for almost all regions, the standard deviation was well below 50% for both HABs and MABs (in both patients and controls), see Additional file 3. This indicates that plasma input-derived BPND can be used as outcome parameter for these larger brain regions. Like VT, regional BPND was on average 1.6 times higher in HAB than in MAB subjects (Fig. 5). For the regions shown in Fig. 5, this difference was statistically significant for frontal cortex (p = 0.001), cingulate cortex (p = 0.006), thalamic grey matter (p = 0.002) and brainstem white matter (p = 0.004). In contrast to the VT findings, regional BPND in both grey and white matter of HAB patients was higher than in HAB healthy controls. This difference was statistically significant for cingulate cortex (p = 0.034), cerebellar white matter (p = 0.034) and non-lesional white matter (p = 0.034). This difference was not observed in the MAB group.

Binding potential for different regions of interest. For high-affinity binders, a higher binding potential (BPND) was seen in multiple sclerosis patients (MS-HAB) than in healthy controls (HC-HAB) for different regions of interest. This difference was not seen in medium-affinity binding (MAB) subjects
Multiple sclerosis lesions
In both HAB and MAB patients with MS, MRI-defined T2 white matter lesions showed a significant increase in BPND compared with non-lesional white matter of the same subject (p = 0.017) (Fig. 6a). There was a strong correlation between the BPND values, except for one patient. When including all the subjects, the correlation coefficient (R2) was 0.246 (slope 1.04 ± 0.744), excluding the one outlier improved the R2 to 0.861 (slope 2.39 ± 0.429). For VT, values were similar for lesions and non-lesional white matter of each MS patient, with an R2 of 0.844 and slope 1.02 ± 0.178 (Fig. 6b).

Kinetic parameter values for the multiple sclerosis lesions. Independent of binding status, a binding potential (BPND) identified increased [18F]DPA714 binding in MS-specific brain lesions compared with non-lesional white matter in the same patients, b whereas volume of distribution (VT) did not depict this difference
Discussion
This proof of concept study in patients with progressive MS showed that quantitative assessment of the second-generation TSPO radiotracer [18F]DPA714 can identify an increased binding potential not only in MS-specific lesions, but for HAB patients also in grey matter and non-lesional white matter. Although the results from this study need to be replicated in a larger cohort, they suggest that [18F]DPA714-PET could be used as an in vivo imaging technique for measuring neuroinflammation in progressive MS.
Neuroinflammation in MS is characterised by complex and dynamic processes, including the activation of microglia [6, 7]. This is most distinct in active MS lesions, identified by gadolinium enhancement on T1-weighted MRI, and chronic MS lesions as seen on T2-weighted MRI [3]. In line with this, an increase in [18F]DPA714 BPND was seen in the focal lesions compared with non-lesional white matter for all patients except one, independent of the patient’s genetic binding status (HAB or MAB). As MRI-defined lesions for these patients with progressive MS were almost exclusively non-enhancing chronic lesions, this increase TSPO binding indicates that these are chronic active lesions [41]. The classification of different stages of MS lesions, such as chronic active and chronic inactive lesions, results from pathological studies. Current conventional MRI techniques can identify active inflammatory lesions, with the use of gadolinium-enhanced T1-weighted scans; however, they cannot further differentiate non-enhancing lesions. Results from the present study suggest that [18F]DPA714 PET can identify different stages of MS lesions in vivo, which could be clinically relevant, e.g. in relation to treatment options and monitoring.
Even more importantly, increased [18F]DPA714 BPND was also seen in non-lesional brain parenchyma, both in white and grey matter, for HAB patients. In relapsing-remitting MS, neuroinflammation is evident from the development of new focal (gadolinium enhancing) lesions, but in primary and secondary progressive MS, neuroinflammation is often more diffuse and low-grade [5]. Therefore, it is highly relevant that a biomarker of neuroinflammation in MS demonstrates not only focal changes, but also more diffuse pathology. As [18F]DPA714 BPND can identify both, this marker of TSPO expression can be used as a biomarker to further investigate neuroinflammation in MS in vivo.
As plasma input-derived BPND was estimated reliably using the 2T4k_VB model, BPND should be preferred over the less specific VT. As VT contains both free, non-specific and specific signals, it is an inferior outcome measured compared with BPND (=k3/k4), which only reflects specific binding. Contrary to BPND analysis, group analysis using VT showed no difference between patients and controls for both HAB and MAB subjects. In addition, VT appeared to be almost similar between different ROIs within each subject, resulting from a nearly homogeneous distribution of DPA714 throughout the brain of an individual. As VT includes the large non-displaceable compartment of [18F]DPA714, this effectively obscured any changes in the disease-specific neuroinflammatory signal. This is also seen in the analysis of the T2 MS lesions in Fig. 6, illustrating that the increase in specific [18F]DPA714 signal in MS lesions depicted by BPND is diluted in the VT values due to the non-displaceable component.
Interestingly, VT was strongly correlated with K1. Although a change in K1 can be related to neuroinflammation, as K1 is a function of blood flow and vascular permeability, a correlation between VT and K1 was unexpected as VT should be independent of K1. A possible explanation for this finding could be related to binding of [18F]DPA714 to the endothelium of blood vessels in the brain [42, 43]. However, the model incorporating a vascular binding component still provided correlated K1 and VT values (Fig. 4b).
Although the 2T4k_VB_1T1k model was preferred according to AIC, it decreased the reliability of the parameter estimates compared with the simpler 2T4k_VB model. Moreover, Kb estimates were inaccurate for many regions, even in subjects where this model was preferred. A proposed method of decreasing the number of parameters, attempting to increase their reliability, is fixing VB to 5%. However, due to the large inter- and intra-subject variation in VB, this seems inappropriate.
Although we used the simpler model in our analysis, the results are similar, as the regional VT values for the two models correlate.
In line with other TSPO studies, assessment of the rs6971 polymorphism defined binding status is important also for quantitative analysis of [18F]DPA714. Although observed differences between HABs and MABs were smaller than those observed for some other second-generation TSPO tracers [24], both mean VT and BPND values were on average 1.6 times higher for HABs compared with MABs for the different brain regions. This indicates that the selection of subjects based on binding status is essential. Furthermore, group differences between HABs were only observed when using BPND, suggesting that [18F]DPA714 in MS could best be studied in HABs only. Due to basal expression of TSPO, the contrast for [18F]DPA714 appears to be too low for MAB subjects to identify patients from controls. Strikingly, the increase in BPND in MS lesions was also seen in MAB patients, indicating that [18F]DPA714 can identify neuroinflammation in MAB subjects in MS lesions, but due to high non-specific binding of the tracer, it is not evident in other ROIs with more low-grade neuroinflammation. As only approximately half of the general population is HAB, this is a limitation of second-generation TSPO studies.
This study is limited mainly by its small sample size. Because of the limited number of subjects in each group, only limited statistical analysis was performed. Nevertheless, based on the promising results of this pilot project, further studies using a larger sample size are warranted. This would allow for evaluation of possible correlations between [18F]DPA714 BPND and clinical outcome scores, such as Expanded Disability Status Scale and Symbol Digit Modalities Test. Finally, assessment of a possible relationship with MRI-derived outcome measures, such as atrophy of cortical and deep grey matter structures, would be of great interest in understanding progressive MS.
Conclusions
Plasma input-derived BPND can reliably be estimated for [18F]DPA714, and a 2T4k_VB model [18F]DPA714 BPND can identify increased neuroinflammation in both grey and non-lesional white matter of patients with progressive MS, with a further increase in T2 MS lesions. As these differences cannot be seen using VT due to inclusion of non-displaceable uptake, BPND is recommended for analysis of [18F]DPA714 studies in patients with MS. Similar to other second-generation TSPO tracers, the effect of genetically defined binding status limits the clinical use of [18F]DPA714. Nevertheless, further studies are warranted to confirm the results of the present proof of concept study.
Abbreviations
- 1T2k:
-
Single-tissue compartment model
- 2T3k:
-
Irreversible two-tissue compartment model
- 2T4k:
-
Reversible two-tissue compartment model
- BPND :
-
Binding potential (k3/k4)
- FLAIR:
-
Fluid Attenuated Inversion Recovery
- HAB:
-
High-affinity binder
- kb :
-
Vascular binding component
- LAB:
-
Low-affinity binder
- MAB:
-
Medium-affinity binder
- MRI:
-
Magnetic resonance imaging
- MS:
-
Multiple sclerosis
- PET:
-
Positron emission tomography
- ROI:
-
Region of interest
- TAC:
-
Time activity curve
- TSPO:
-
18-kDa translocator-protein
- V B :
-
Blood volume parameter
- V T :
-
Volume of distribution
References
O'Connor P, Comi G, Freedman MS, Miller AE, Kappos L, Bouchard JP, et al. Long-term safety and efficacy of teriflunomide: nine-year follow-up of the randomized TEMSO study. Neurology. 2016;86:10.
Rovira A, Wattjes MP, Tintore M, Tur C, Yousry TA, Sormani MP, et al. Evidence-based guidelines: MAGNIMS consensus guidelines on the use of MRI in multiple sclerosis-clinical implementation in the diagnostic process. Nat Rev Neurol. 2015;11:8.
Wattjes MP, Rovira A, Miller D, Yousry TA, Sormani MP, de Stefano MP, et al. Evidence-based guidelines: MAGNIMS consensus guidelines on the use of MRI in multiple sclerosis--establishing disease prognosis and monitoring patients. Nat Rev Neurol. 2015;11:10.
Barkhof F. The clinico-radiological paradox in multiple sclerosis revisited. Curr Opin Neurol. 2002;15:3.
Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol. 2012;8:11.
Hemmer B, Kerschensteiner M, Korn T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 2015;14:4.
Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14:2.
Airas L, Rissanen E, Rinne J. Imaging of microglial activation in MS using PET: research use and potential future clinical application. Mult Scler. 2017;23:4.
Hagens M, van Berckel B, Barkhof F. Novel MRI and PET markers of neuroinflammation in multiple sclerosis. Curr Opin Neurol. 2016;29:3.
Cumming P, Burgher B, Patkar O, Breakspear M, Vasdev N, Thomas P, et al. Sifting through the surfeit of neuroinflammation tracers. J Cereb Blood Flow Metab. 2018;38:2.
Vowinckel E, Reutens D, Becher B, Verge G, Evans A, Owens T, et al. PK11195 binding to the peripheral benzodiazepine receptor as a marker of microglia activation in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neurosci Res. 1997;50:2.
Banati RB, Newcombe J, Gunn RN, Cagnin A, Turkheimer F, Heppner F, et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain. 2000;123(Pt 11):2321–37.
Politis M, Giannetti P, Su P, Turkheimer F, Keihaninejad S, Wu K, et al. Increased PK11195 PET binding in the cortex of patients with MS correlates with disability. Neurology. 2012;79:6.
Rissanen E, Tuisku J, Rokka J, Paavilainen T, Parkkola R, Rinne JO, et al. In vivo detection of diffuse inflammation in secondary progressive multiple sclerosis using PET imaging and the radioligand 11C-PK11195. J Nucl Med. 2014;55:6.
Giannetti P, Politis M, Su P, Turkheimer F, Malik O, Keihaninejad S, et al. Microglia activation in multiple sclerosis black holes predicts outcome in progressive patients: an in vivo [11C](R)-PK11195-PET pilot study. Neurobiol Dis. 2014;65:203–10.
Giannetti P, Politis M, Su P, Turkheimer FE, Malik O, Keihaninejad S, et al. Increased PK11195-PET binding in normal-appearing white matter in clinically isolated syndrome. Brain. 2015;138:Pt 1.
Boutin H, Chauveau F, Thominiaux C, Gregoire MC, James ML, Trebossen R, et al. 11C-DPA-713: a novel peripheral benzodiazepine receptor PET ligand for in vivo imaging of neuroinflammation. J Nucl Med. 2007;48:4.
Fookes CJ, Pham TQ, Mattner F, Greguric I, Loc'h C, Liu X, et al. Synthesis and biological evaluation of substituted [18F]imidazo[1,2-a]pyridines and [18F]pyrazolo[1,5-a]pyrimidines for the study of the peripheral benzodiazepine receptor using positron emission tomography. J Med Chem. 2008;51:13.
Kreisl WC, Fujita M, Fujimura Y, Kimura N, Jenko KJ, Kannan P, et al. Comparison of [11C]-(R)-PK 11195 and [11C]PBR28, two radioligands for translocator protein (18 kDa) in human and monkey: implications for positron emission tomographic imaging of this inflammation biomarker. NeuroImage. 2010;49:4.
Wadsworth H, Jones PA, Chau WF, Durrant C, Fouladi N, Passmore J, et al. [18F]GE-180: a novel fluorine-18 labelled PET tracer for imaging Translocator protein 18 kDa (TSPO). Bioorg Med Chem Lett. 2012;22:3.
Zhang MR, Maeda J, Ogawa M, Noguchi J, Ito T, Yoshida Y, et al. Development of a new radioligand, N-(5-fluoro-2-phenoxyphenyl)-N-(2-[18F]fluoroethyl-5-methoxybenzyl)acetamide, for pet imaging of peripheral benzodiazepine receptor in primate brain. J Med Chem. 2004;47:9.
James ML, Fulton RR, Vercoullie J, Henderson DJ, Garreau L, Chalon S, et al. DPA-714, a new translocator protein-specific ligand: synthesis, radiofluorination, and pharmacologic characterization. J Nucl Med. 2008;49:5.
Peyronneau MA, Saba W, Goutal S, Damont A, Dolle F, Kassiou M, et al. Metabolism and quantification of [18F]DPA-714, a new TSPO positron emission tomography radioligand. Drug Metab Dispos. 2013;41:1.
Owen DR, Gunn RN, Rabiner EA, Bennacef I, Fujita M, Kreisl WC, et al. Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J Nucl Med. 2011;52:1.
Lavisse S, Garcia-Lorenzo D, Peyronneau MA, Bodini B, Thiriez C, Kuhnast B, et al. Optimized quantification of translocator protein radioligand 18F-DPA-714 uptake in the brain of genotyped healthy volunteers. J Nucl Med. 2015;56:7.
Golla SS, Boellaard R, Oikonen V, Hoffmann A, van Berckel BN, Windhorst AD, et al. Quantification of [18F]DPA-714 binding in the human brain: initial studies in healthy controls and Alzheimer’s disease patients. J Cereb Blood Flow Metab. 2015;35:5.
Ribeiro MJ, Vercouillie J, Debiais S, Cottier JP, Bonnaud I, Camus V, et al. Could 18F-DPA-714 PET imaging be interesting to use in the early post-stroke period? EJNMMI Res. 2014;4:28.
Hamelin L, Lagarde J, Dorothee G, Leroy C, Labit M, Comley RA, et al. Early and protective microglial activation in Alzheimer’s disease: a prospective study using 18F-DPA-714 PET imaging. Brain. 2016;139:Pt 4.
Salinas CA, Searle GE, Gunn RN. The simplified reference tissue model: model assumption violations and their impact on binding potential. J Cereb Blood Flow Metab. 2015;35:2.
Folkersma H, Boellaard R, Vandertop WP, Kloet RW, Lubberink M, Lammertsma AA, et al. Reference tissue models and blood-brain barrier disruption: lessons from (R)-[11C]PK11195 in traumatic brain injury. J Nucl Med. 2009;50:12.
Boellaard R, van Lingen A, van Balen SC, Hoving BG, Lammertsma AA. Characteristics of a new fully programmable blood sampling device for monitoring blood radioactivity during PET. Eur J Nucl Med. 2001;28:1.
Steenwijk MD, Pouwels PJ, Daams M, van Dalen JW, Caan MW, Richard E, et al. Accurate white matter lesion segmentation by k nearest neighbor classification with tissue type priors (kNN-TTPs). Neuroimage Clin. 2013;3:462–9.
Chard DT, Jackson JS, Miller DH, Wheeler-Kingshott CA. Reducing the impact of white matter lesions on automated measures of brain gray and white matter volumes. J Magn Reson Imaging. 2010;32:1.
Cizek J, Herholz K, Vollmar S, Schrader R, Klein J, Heiss WD. Fast and robust registration of PET and MR images of human brain. NeuroImage. 2004;22:1.
Vollmar S, Cizek J, Sue J, Klein A, Jacbos K, Herholz K. VINCI-volume imaging in neurological research, co-registration and ROIs included. Forschung und wissenschaftliches Rechnen 2003 (Kremer K, Macho V, eds); 2004.
Svarer C, Madsen K, Hasselbalch SG, Pinborg LH, Haugbol S, Frokjaer VG, et al. MR-based automatic delineation of volumes of interest in human brain PET images using probability maps. NeuroImage. 2005;24:4.
Hammers A, Allom R, Koepp MJ, Free SL, Myers R, Lemieux L, et al. Three-dimensional maximum probability atlas of the human brain, with particular reference to the temporal lobe. Hum Brain Mapp. 2003;19:4.
Frisoni GB, Boccardi M, Barkhof F, Blennow K, Cappa S, Chiotis K, et al. Strategic roadmap for an early diagnosis of Alzheimer’s disease based on biomarkers. Lancet Neurol. 2017;16:8.
Rizzo G, Veronese M, Tonietto M, Zanotti-Fregonara P, Turkheimer FE, Bertoldo A. Kinetic modeling without accounting for the vascular component impairs the quantification of [11C]PBR28 brain PET data. J Cereb Blood Flow Metab. 2014;34:6.
Wimberley C, Lavisse S, Brulon V, Peyronneau MA, Leroy C, Bodini B, et al. Impact of endothelial TSPO on the quantification of 18F-DPA-714. J Nucl Med. 2018;59:307–14.
van der Valk P, De Groot CJ. Staging of multiple sclerosis (MS) lesions: pathology of the time frame of MS. Neuropathol Appl Neurobiol. 2000;26:1.
Tomasi G, Edison P, Bertoldo A, Roncaroli F, Singh P, Gerhard A, et al. Novel reference region model reveals increased microglial and reduced vascular binding of 11C-(R)-PK11195 in patients with Alzheimer’s disease. J Nucl Med. 2008;49:8.
Turkheimer FE, Edison P, Pavese N, Roncaroli F, Anderson AN, Hammers A, et al. Reference and target region modeling of [11C]-(R)-PK11195 brain studies. J Nucl Med. 2007;48:1.
Acknowledgements
The authors would like to thank Mattia Veronese (Department of Neuroimaging, King’s College London) for his help on the implementation of the kinetic model including a vascular binding component.
Funding
This research has received funding from Amsterdam Neuroscience and the Dutch MS Research Foundation (programme grant 14-358e). Both organisations had no role in the design of the study or the collection, analysis and interpretation of data.
Availability of data and materials
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
The study was designed by MH, AMvD, JK, ADW, FB and BvB. Tracer development was done by TAR, MK and ADW. Data were collected by MH, MTW, PS and JB. Analysis of the data was performed by MH, SVG, MY, DH, MDS, RCS and AAL and data were interpreted by MH, SVG, MY, BvB and AAL. The manuscript was drafted by MH and SVG. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Medical Ethics Review Board of the VU University Medical Center and was registered under the number 2014.356. All subjects gave written informed consent prior to participation.
Consent for publication
Not applicable.
Competing interests
DH is employed by Philips Healthcare. JK has accepted speaker and consultancy fees from Merck-Serono, Teva, Biogen, Genzyme, Roche and Novartis. FB is supported by the NIHR UCLH Biomedical Research Centre and serves as a consultant for Bayer-Schering Pharma, Sanofi-Aventis, Genzyme, Biogen-Idec, Teva, Novartis, Roche, Synthon BV and Jansen Research. All other authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
2T4k_VB_1T1k - Percentage standard deviation for BPND (k3/k4). Additional file provides the percentage of standard deviation for the for 2T4k_VB_1T1k BPND (k3/k4) for the different large regions of interest. (PDF 106 kb)
Additional file 2:
2T4k_VB_1T1k - Percentage standard deviation for kb. Additional file provides the percentage of standard deviation for the for 2T4k_VB_1T1k kb for the different large regions of interest. (PDF 106 kb)
Additional file 3:
2T4k_VB - Percentage standard deviation for BPND (k3/k4). Additional file provides the percentage of standard deviation for the arterial input-derived BPND (k3/k4) for the different large regions of interest. (PDF 106 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hagens, M.H.J., Golla, S.V., Wijburg, M.T. et al. In vivo assessment of neuroinflammation in progressive multiple sclerosis: a proof of concept study with [18F]DPA714 PET. J Neuroinflammation 15, 314 (2018). https://doi.org/10.1186/s12974-018-1352-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-018-1352-9