
Abstract
Background
Neuroinflammation is a crucial factor contributing to neurological injuries after intracerebral hemorrhage (ICH). C1q/TNF-related protein 9 (CTRP9), an agonist of adiponectin receptor 1 (AdipoR1), has recently been shown to reduce inflammatory responses in systemic diseases. The objective of this study was to investigate the protective role of CTRP9 against neuroinflammation after ICH in a mouse model and to explore the contribution of adenosine monophosphate-activated protein kinase (AMPK)/nuclear factor kappa B (NFκB) pathway in AdipoR1-mediated protection.
Methods
Adult male CD1 mice (n = 218) were randomly assigned to different groups for the study. ICH was induced via intrastriatal injection of bacterial collagenase. Recombinant CTRP9 (rCTRP9) was administered intranasally at 1 h after ICH. To elucidate the underlying mechanism, AdipoR1 small interfering ribonucleic acid (siRNA) and selective phosphorylated AMPK inhibitor Dorsomorphin were administered prior to rCTRP9 treatment. Brain edema, short- and long-term neurobehavior evaluation, blood glucose level, western blot, and immunofluorescence staining were performed.
Results
Endogenous CTRP9 and AdipoR1 expression was increased and peaked at 24 h after ICH. AdipoR1 was expressed by microglia, neurons, and astrocytes. Administration of rCTRP9 reduced brain edema, improved short- and long-term neurological function, enhanced the expression of AdipoR1 and p-AMPK, and decreased the expression of phosphorylated NFκB and inflammatory cytokines after ICH. The protective effects of rCTRP9 were abolished by administration of AdipoR1 siRNA and Dorsomorphin.
Conclusions
Our findings demonstrated that administration of rCTRP9 attenuated neuroinflammation through AdipoR1/AMPK/NFκB signaling pathway after ICH in mice, thereby reducing brain edema and improving neurological function after experimental ICH in mice. Therefore, CTRP9 may provide a potential therapeutic strategy to alleviate neuroinflammation in ICH patients.
Similar content being viewed by others


Background
Spontaneous intracerebral hemorrhage (ICH) is a severe type of stroke with high morbidity and mortality, accounting for about 15% of all stroke patients [1]. Intracerebral hematoma oppresses surrounding brain tissue is considered primary brain injury. Then, red blood cell debris and blood components breakdown can initiate the secondary brain injury and persist for several days to weeks in the perihematoma area [2]. Increasing evidence indicates that inflammatory response that occurs in the early stage after ICH is a key factor leading to secondary brain injury induced by ICH. Treatment to inhibit neuroinflammation after the onset of ICH can provide critical therapeutic strategy to improve outcomes after ICH.
Adiponectin is a major adipocyte-derived hormone that exerts its beneficial physiological effects such as regulation of metabolism, anti-atherosclerosis, and anti-inflammatory effects by activating its receptors [3,4,5]. Adiponectin receptor 1 (AdipoR1) is the main isoform of adiponectin receptors. Activation of AdipoR1 has been shown to have anti-inflammatory effects in the brain [6]. The C1q/TNF-related protein (CTRP) family is a novel analog of adiponectin, among which the globular C1q domain of CTRP9 has the highest degree of amino acid sequence homology with adiponectin. Studies have shown that CTRP9 has a protective effect against inflammation after cardiac ischemia [7, 8]. However, its role in stroke has not been investigated.
Adenosine monophosphate-activated protein kinase (AMPK) signaling system plays a key role in cellular and organismal survival by its ability to maintain metabolic homeostasis. Activation of AMPK has been shown to inhibit inflammatory response in various injury models while suppression of AMPK activity was associated with increased inflammation [9, 10]. Furthermore, the nuclear factor kappa B (NFκB) signaling system is one of the primary pathways involved in pro-inflammatory responses. Previous studies have shown that activation of AMPK signaling can downregulate the function of NFκB system [11,12,13]. To date, the anti-inflammatory role of CTRP9 in the brain has not been studied.
In the present study, we hypothesized the following three points: (1) rCTRP9 administration by intranasal route improves neurological deficits and brain edema after ICH; (2) rCTRP9 administration attenuates the production of inflammatory mediators following ICH; (3) the anti-inflammatory effect of rCTRP9 is mediated through the AdipoR1/AMPK/NFκB pathway after ICH.
Methods
Animals
Eight-week-old male CD1 mice (weight 30–40 g, Charles River, Wilmington, MA) were housed in a 12-h light/dark cycle at a controlled temperature and humidity with unlimited access to food and water. All experimental procedures were performed following the study protocol approved by the Institutional Animal Care and Use Committee (IACUC) at Loma Linda University in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Experimental design
In the present study, all mice were randomly assigned to the following experiments. The following five separate experiments are shown in the timeline of experimental design (Additional file 1: Figure S1). The summary of experimental groups, animal numbers, and mortality rate in the study are listed in a table (Additional file 2: Table S1).
Experiment 1
To evaluate time course expression of endogenous CTRP9 and AdipoR1 after ICH, 36 mice were randomly divided into 6 groups for western blot: sham, ICH after 3, 6, 12, 24, and 72 h (n = 6/group). Additional two mice were used for immunofluorescence staining at 24 h post-ICH. Western blot was used to detect the expression of CTRP9 and AdipoR1 in the ipsilateral (right) hemisphere. Immunofluorescence staining was performed to localize AdipoR1 on neurons, astrocytes, and microglia.
Experiment 2
To determine the best treatment dosage for rCTRP9, 30 mice were randomly divided into 5 groups, n = 6/group: sham, ICH + vehicle (PBS), ICH + rCTRP9 (0.03 μg/g), ICH + rCTRP9 (0.1 μg/g), ICH+ rCTRP9 (0.3 μg/g). rCTRP9 was administered intranasally at 1 h post-ICH. Neurobehavioral tests and brain water content (BWC) were assessed at 24 h post-ICH. Blood glucose level was measured starting 1, 2, 3, 6, and 24 h after ICH.
Experiment 3
To determine the presence of rCTRP9 in the brain after intranasal administration, mice (n = 24) were divided into 4 groups, n = 6/group: naive, naïve+rCTRP9 (0.1 μg/g), ICH, ICH + rCTRP9 (0.1 μg/g). Based on results from experiment 2, medium dosage (0.1 μg/g) of rCTRP9 was chosen for the study. Neurobehavioral tests and western blot to measure rCTRP9 expression in ipsilateral (right) hemisphere were performed at 24 h post-ICH. Next, for the effects of the drug at 72 h after ICH, 18 mice were divided into 3 groups, n = 6/group: sham, ICH + vehicle (PBS), ICH + rCTRP9 (0.1 μg/g). Neurobehavioral tests and BWC were evaluated at 72 h post-ICH.
Experiment 4
To assess long-term neurobehavioral function, 24 mice were divided into 3 groups: sham, ICH + vehicle (PBS), ICH + rCTRP9 (0.1 μg/g), n = 8/group. The foot fault test and rotarod test were performed on days 7, 14, and 21 post-ICH. Morris water maze test was conducted on days 21–25 post-ICH.
Experiment 5
To verify the efficacy of AdiporR1 siRNA to knockdown the expression of AdipoR1, 24 mice were divided into 4 groups, n = 6/group: naive, naïve+AdipoR1 small interfering ribonucleic acid (siRNA), ICH, ICH + AdipoR1 siRNA. Next, to verify the anti-inflammatory mechanism of rCTRP9, mice (n = 42) were divided into seven groups: sham, ICH + vehicle (PBS), ICH + rCTRP9 (0.1 μg/g), ICH + rCTRP9 + AdipoR1 siRNA, ICH + rCTRP9 + scramble siRNA (Scr siRNA), ICH + rCTRP9 + Dorsomorphin, ICH + rCTRP9 + DMSO (5 μl of 5% dimethyl sulfoxide in PBS), n = 6/group. Neurobehavioral tests and western blot were performed at 24 h post-ICH.
ICH model
ICH was induced by intrastriatal injection of bacterial collagenase into the right basal ganglia as previously described [14]. Briefly, mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) (2:1 vol/vol, intraperitoneal injection) and positioned prone in a stereotaxic head frame. Bacterial collagenase (0.075 units, type VII-S; Sigma-Aldrich, St. Louis, MO) was dissolved in 0.5 μL PBS and infused into the right basal ganglia at a rate of 0.1667 μL/min, using an infusion pump (Stoelting, IL). The needle was left for an additional 5 min to prevent possible leakage of the collagenase solution, and withdrawn slowly at a rate of 1 mm/min. After removal of the needle, the cranial burr hole was sealed with bone wax, the incision was sutured, and 0.4 mL of normal saline was injected subcutaneously, and the mice were allowed to recover. The sham operation was performed with PBS only. All ICH animals with hematoma and neurological deficits were included. Animals that died before experimental endpoints reached were replaced with new animals. Animals with no hematoma were excluded.
Drug administration
Recombinant CTRP9 (rCTRP9) (Novus, CO) dissolved in PBS was administered intranasally at 1 h post-ICH as previously reported [15]. Briefly, rCTRP9 was diluted in PBS, and three different doses of rCTRP9 (0.03, 0.1, and 0.3 μg/g) were treated. Nasal administration of rCTRP9 was performed at 1 h after ICH induction. A total volume 20 μL of rCTRP9 was administered intranasally. Mice, still under anesthesia, were placed in a supine position, and rCTRP9 was administered alternating drops (5 μL/drop) every 2 min between the left and right nares over a period of 20 min. The ICH + vehicle group received an equal volume of phosphate-buffered saline (PBS).
Intracerebroventricular injection
AdipoR1 siRNA and scramble siRNA (scr siRNA) (Life Technologies, NY) were delivered via intracerebroventricular (ICV) injection 48 h before ICH as previously described [16]. Briefly, AdipoR1 SiRNA or scr siRNA (100 pmol/mouse) was dissolved in sterile RNAs free resuspension buffer according to the manufacturer’s instructions (Life Technologies, NY). The ICV injections into the left ventricle were performed using the following coordinates relative to bregma (0.22 mm posterior, 1.0 mm lateral, and 2.25 mm deep) at a rate of 0.667 μL/min at 48 h prior to surgery. After waiting for 5 min, the needle was removed over a period of 3 min. The burr hole was sealed with bone wax. Dorsomorphin (5 μg/mouse, Sigma, MO) was dissolved in DMSO and given by ICV injection into left ventricle as described above at 30 min before ICH surgery.
Short-term neurobehavior assessment
Short-term neurobehavioral tests were performed with modified Garcia score test, forelimb placement test, and corner turn test by an experienced investigator who was blinded to the experimental groups at 24 and 72 h post-ICH, as previously reported [17]. Garcia score assessment with a 21-point score evaluating spontaneous activity, axial sensation, vibrissae proprioception, symmetry of limb movement, lateral turning, forelimb walking, climbing, and grabbing was conducted. The forelimb placement test was used to investigate the animals’ responsiveness to vibrissae stimulation; the placement of the left and right forelimbs was investigated for 10 consecutive trials. Then, left forelimb placement was calculated as left forelimb placement/(left forelimb placement + right forelimb placement) × 100%. For the corner turn test, animals were allowed to advance into a 30° corner and exit by turning either to the left or right. Choice of turning was recorded for a total of 10 trials, and a score was calculated as number of left turns/all trials × 100.
Brain water content measurement
The brain water content (BWC) was measured as previously reported using the wet/dry method [17]. Mice were euthanized under deep anesthesia and the brains were removed immediately and divided into five parts: ipsilateral and contralateral cortex, ipsilateral and contralateral basal ganglia, and cerebellum. All brain samples were weighed on analytical microbalance (APX-60 Denver Instrument, NY) to obtain the wet weight (WW). Then the samples were dried at 100 °C for 48 h to obtain the dry weight (DW). Brain water content (%) was calculated using the following formula: (WW − DW)/WW*100%.
Blood glucose measurement
Plasma glucose level was measured by blood glucose meter (Ascensia Diabetes Care US Inc., NJ) in the blood collected from tail vein before ICH and at 1, 2, 3, 6, and 24 h after ICH in each group.
Long-term neurobehavior assessment
Foot-fault and rotarod test were performed at the first, second, and third weeks post-ICH to assess sensorimotor function, coordination, and balance as previously described [18]. Water maze tests, including swim distance and escape latency, were performed at days 21 to 25 post-ICH to evaluate memory and spatial learning as previously described [19]. Morris water maze test probe quadrant duration was evaluated on day 25 post-ICH.
Western blot analysis
Western blotting was performed as described previously [20]. Briefly, the brain protein samples were prepared using Ripa Lysis buffer (Bio-Rad, CA), and equal amounts of protein were run on an SDS-PAGE gel. After being electrophoresed and transferred to a nitrocellulose membrane, the membrane was blocked and incubated with primary antibody overnight at 4 °C. The following primary antibodies were used: AdipoR1 (1:1000, ab126611), p-AMPKα (1:1000, ab133448), AMPKα (1:1000, ab32047), p-NFκB (1:1000, ab86299), NFκB (1:2000, ab16502), tumor necrosis factorα (TNFα) (1:1000, ab6671), interleukin-6 (IL-6) (1:1000, ab6672) (all from Abcam, MA), CTRP9 (1:500, NBP2–46834, Novus, CO), and adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1 (APPL1) (1:1000, sc-271,901, Santa Cruz Biotechnology, CA). β-actin was used as an internal loading control. The respective secondary antibodies were incubated for 1 h at room temperature. The bands were probed with an ECL Plus chemiluminescence reagent Kit (Amersham Biosciences, Arlington Heights, PA) and visualized with the image system (Bio-Rad, Versa Doc, model 4000). Relative density of the protein immunoblot images were analyzed by ImageJ software (ImageJ 1.4, NIH, USA).
Immunofluorescence staining
At 24 h following ICH, mice were perfused under deep anesthesia with cold PBS (pH 7.4). Mice were then infused with 10% formalin. Brains were then removed and fixed in formalin at 4 °C for a minimum of 3 days. Samples were then dehydrated with 30% sucrose in PBS (pH 7.4). The frozen coronal slices (8 μm thick) were sectioned in cryostat (CM3050S; Leica Microsystems). Double immunofluorescence staining was performed as previously described [20]. The primary antibodies included anti-AdipoR1 (1:200, ab126611), anti-Iba-1 (1:200, ab178847), anti-NeuN (1:200, ab177487), and anti-GFAP (1:200, ab16997) (all from Abcam, MA).
Statistical analysis
All animals were randomly assigned to different groups. All the experimental tests were blinded to the surgeons and researchers who did the experiments and analyzed the research data. All tests for exploratory studies were performed two-sided. All data were expressed as the mean and standard deviation (mean ± SD). Statistical analysis was performed with GraphPad Prism (Graph Pad Software, San Diego, CA). One-way analysis of variance (ANOVA) is followed by multiple comparisons between groups using Tukey’s post hoc test. Two-way repeated measures ANOVA was used to analyze the long-term neurobehavioral functions over time. Statistical significance was set at p < 0.05.
Results
Animal mortality rate
A total of 218 mice were used of which 56 were sham and 162 mice underwent ICH induction. None of the sham mice died, and the mortality rate in ICH group was 9.2% (15/162). Three mice were excluded from the study because of no hemorrhage. An additional file shows this in more details (Additional file 2: Table S1).
Endogenous CTRP9 and AdipoR1 expression were increased after ICH
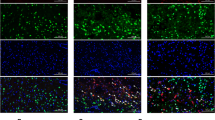
The expression of endogenous CTRP9 and AdipoR1 was increased after ICH with a peak at 24 h compared to sham group and then decreased at 72 h after ICH (p < 0.05, Fig. 1a, b). Immunofluorescence staining showed that AdipoR1 was expressed in microglia (Iba-1), neurons (NeuN), and astrocytes (GFAP) at 24 h post-ICH (Fig. 1c).

Expression of C1q/TNF-related protein 9 (CTRP9) and adiponectin receptor 1 (AdipoR1) after intracerebral hemorrhage (ICH). a Representative western blot images and quantitative analyses of CTRP9 time course after ICH. b Representative western blot images and quantitative analyses of AdipoR1 time course after ICH. Values are expressed as mean ± SD. *p < 0.05, **p < 0.01 vs. sham group. N = 6. c Double immunofluorescence staining for AdipoR1 (red) in microglia (Iba-1, green), neurons (NeuN, green), and astrocytes (GFAP, green) in right basal cortex 24 h after ICH. Scale bar = 50 μm. N = 2. DAPI, 4′,6-diamidino-2-phenylindole; Iba-1, ionized calcium binding adaptor molecule-1; NeuN, neuronal nuclear; GFAP, glial fibrillary acidic protein
rCTRP9 attenuated neurobehavioral deficits and reduced brain edema at 24 h after ICH
The neurobehavioral score was significantly decreased in vehicle group compared with the sham group at 24 h post-ICH. Administration of medium dose and high dose of rCTRP9 significantly improved neurological function and decreased BWC in the right basal ganglia and cortex compared with ICH + vehicle group (p < 0.05, Fig. 2a–d).

The effects of different doses of rCTRP9 on neurobehavior tests, brain water content (BWC), and blood glucose at 24 h post-ICH. a Modified Garcia test, b forelimb placement test, c corner turn test, and d BWC at 24 h post-ICH. e Blood glucose levels measured at 0, 1, 2, 3, 6, and 24 h post-ICH. Values are expressed as mean ± SD. *p < 0.05, **p < 0.01 vs. sham group; #p < 0.05 vs. ICH + vehicle group; @p < 0.05 vs. before ICH (0 h); N = 6
rCTRP9 did not change blood glucose level after ICH
Blood glucose level increased significantly at 1 h post-surgery in all groups compared to baseline before surgery (p < 0.05), then continued to decline and reached nadir at 24 h post-ICH (Fig. 2e). There was no significant difference in blood glucose levels between experimental groups when compared at the same time point (p > 0.05). Based on above results, medium dosage rCTRP9 was used in the following experiments.
Nasal rCTRP9 administration increased the brain expression level of CTRP9 in naïve and ICH mice
At 24 h after nasal administration of rCTRP9, the expression of CTRP9 in naïve+CTRP9 group was significantly increased compared with naïve group. The expression level of CTRP9 was significantly increased in ICH + rCTRP9 group compared to ICH group at 24 h after ICH (p < 0.05, Fig. 3a). The neurobehavioral score in the ICH + rCTRP9 group was significantly improved compared with ICH group (p < 0.05, Fig. 3b–d).

Brain expression of CTRP9 and neurological function evaluation after nasal administration of rCTRP9 at 24 h post-ICH. a Representative western blot images and quantitative analyses of CTRP9. Nasal administration of rCTRP9 increased the expression level of CTRP9 in naïve mice and ICH mice at 24 h post-ICH. b Modified Garcia test, c forelimb placement test, and d corner turn test showed that nasal administration of rCTRP9 improved neurological function at 24 h post-ICH. Values are expressed as mean ± SD. *p < 0.05 vs. naive group; #p < 0.05 vs. ICH group; N = 6
rCTRP9 attenuated neurobehavioral deficits and reduced brain edema at 72 h after ICH
The neurobehavioral score was significantly improved, and brain edema in the right basal ganglia and cortex was reduced with rCTRP9 treatment compared with ICH + vehicle group at 72 h after ICH (p < 0.05, Fig. 4a–d).

rCTRP9 improved neurological deficits and decreased brain water content (BWC) at 72 h post-ICH. a Modified Garcia test, b forelimb placement test, c corner turn test, and d BWC at 72 h post-ICH. Values are expressed as mean ± SD. *p < 0.05, **p < 0.01 vs. sham group; #p < 0.05 vs. ICH + vehicle group; N = 6
rCTRP9 improved long-term neurobehavioral outcomes after ICH
In the foot fault test, ICH + vehicle group showed significantly increased foot faults compared to sham group and in the rotarod test, ICH + vehicle group had shorter latency to fall compared to sham group at 1, 2, and 3 weeks after ICH. Animals with rCTRP9 treatment showed significant improvement in both tests (p < 0.05; Fig. 5a, b).

rCTRP9 improved long-term neurobehavioral function after ICH. a Foot fault test and rotarod test on days 7, 14, and 21 after ICH. b Morris water maze indicated by swim distance and escape latency on 21–25 days after ICH. c Morris water maze test probe quadrant duration on day 25 post-ICH. Values are expressed as mean ± SD. *p < 0.05, **p < 0.01 vs. sham group; #p < 0.05 vs. ICH + vehicle group. N = 8
In the water maze test, ICH + vehicle group showed significantly longer swim distance and escape latency. The rCTRP9 treatment group had significantly improved performance on days 3 to 5 of testing (p < 0.05; Fig. 5c, d). In the probe trial, the ICH + vehicle group spent less time in the target quadrant compared with sham group, while rCTRP9 treatment markedly increased the time spent in the probe quadrant (p < 0.05, Fig. 5e).
AdipoR1 siRNA aggravated neurological deficits and decreased AdipoR1 expression in naïve and ICH mice
AdipoR1 siRNA administration significantly decreased the expression of AdipoR1 and reduced neurobehavioral score in naïve+AdipoR1 siRNA group compared with naïve group. Similarly, the expression level of AdipoR1 was significantly decreased, and neurobehavior score was reduced in ICH + AdipoR1 siRNA group compared with ICH group (p < 0.05, Fig. 6a–d).

AdipoR1 expression and neurological function evaluation after AdipoR1 siRNA administration in naïve and ICH mice. a Representative western blot images and quantitative analyses of AdipoR1 expression. AdipoR1 siRNA decreased the expression of AdipoR1 in naïve mice and ICH mice at 24 h post-ICH. b Modified Garcia test, b forelimb placement test, and c corner turn test showed that AdipoR1 siRNA aggravated neurological deficits in naïve mice and ICH mice at 24 h post-ICH. Values are expressed as mean ± SD. *p < 0.05 vs. naive group; #p < 0.05 vs. ICH group; N = 6
AdipoR1 siRNA and Dorsomorphin abolished the anti-inflammatory effects of rCTRP9
AdipoR1 siRNA but not scramble siRNA reversed neurobehavioral improvement observed in ICH + rCTRP9 mice at 24 h post-ICH. Likewise, Dorsomorphin significantly decreased neurobehavioral score compared with ICH + rCTRP9 + DMSO group (p < 0.05, Fig. 7a–c).

Knockdown of AdipoR1 and p-AMPK abolished improvement in neurological function and anti-inflammatory effects of rCTRP9 at 24 h post-ICH. a Modified Garcia test, b forelimb placement test, c corner turn test, and d representative western blot images and quantitative analyses of CTRP9, AdipoR1, APPL1, p-AMPK/AMPK, p-NFκB/NFκB, TNF-α, and IL-6 at 24 h post-ICH. Values are expressed as mean ± SD. *p < 0.05, **p < 0.01 vs. sham group; #p < 0.05, ##p < 0.01 vs. ICH + vehicle group; &p < 0.05 vs. rCTRP9 + Scr siRNA, and @p < 0.05 vs. rCTRP9 + DMSO group. Scr, scramble; siRNA, small interfering RNA. N = 6
The expression of CTRP9 was significantly increased in all ICH groups that received rCTPR9 via intranasal administration compared to sham and ICH + vehicle groups. The expression of AdipoR1, APPL1, and p-AMPK increased after rCTRP9 administration in ICH + rCTRP9 group compared to ICH + vehicle group. AdipoR1 siRNA significantly decreased the expression of AdipoR1, APPL1, and p-AMPK, while increasing the expression of p-NFκB, TNFα, and IL-6 when compared with ICH + rCTRP9 + Scr siRNA group. In addition, Dorsomorphin intervention significantly suppressed p-AMPK expression, while increasing p-NFκB, TNFα, and IL-6 expression when compared with ICH + rCTRP9 + DMSO group (p < 0.05; Fig. 7d).
Discussion
In the present study, we first showed that the expression of CTRP9 and AdipoR1 increased in the brain at 24 h after ICH. Moreover, we found that rCTRP9 administration improved both short- and long-term neurobehavioral outcomes, alleviated brain edema, and attenuated neuroinflammation after ICH, which were accompanied by an increase in AdipoR1 and p-AMPK expression and a decrease in pro-inflammatory factors TNFα and IL-6 expression. Additionally, we determined that AdipoR1/AMPK/NFκB signaling pathway was involved in the anti-inflammatory effects of rCTRP9 after ICH. These findings suggest that rCTRP9 administration possibly improved outcomes after ICH at least in part by attenuating neuroinflammation through AdipoR1/AMPK/NFκB signaling pathway. The schematic mechanism was shown in a figure (Additional file 3: Figure S2).
Adiponectin is a biologically active polypeptide secreted by adipocytes and plays important roles in the regulation of energy homeostasis and insulin sensitivity. However, adiponectin knockout mice showed a relatively modest phenotype in the absence of diet or metabolic stress [21]. These findings suggested that there was a potential effective compensatory mechanism involved. CTRP9 shares the highest degree of sequence identity with adiponectin at the presumed functional globular domain, and has similar biochemical properties to adiponectin [22]. Existing research shows that CTRP9 has a variety of beneficial functions such as relaxing blood vessels, protecting the vascular endothelium, regulating metabolism, and reducing the expression of inflammatory factors [23, 24]. In addition, CTRP9 has also been shown to decrease myocardial infarct size by inhibiting the inflammatory response [25]. However, its role in the brain has never been studied.
In the present study, we demonstrated for the first time that CTRP9 has anti-inflammatory effects in ICH mouse model. The outcomes that we measured in this study have been utilized as standard outcome measurement in previously published ICH rodent model studies in which thrombin inhibition, microglia inactivation, and iron chelation were shown to have similar beneficial effects as observed in our study [26,27,28]. We evaluated the temporal expression of endogenous CTRP9 for 3 days after ICH. The results indicated that CTRP9 expression increased after ICH and reached the highest level at 24 h post-ICH. It has been reported that physiological levels of adiponectin in both human and mouse serum were elevated in response to inflammatory stimuli [29,30,31,32]. Our findings are consistent with a previous study that showed endogenous adiponectin expression significantly increased in cerebral ischemic areas after middle cerebral artery occlusion (MCAO) [33]. Acute injury or stress has been proposed to increase plasma concentration of elastase, which promotes the generation of globular adiponectin from full-length adiponectin [22, 34]. The acute stress following ICH injury may therefore contribute to increase endogenous CTRP9 expression after ICH. In addition, we observed that nasal administration of rCTRP9 increased the brain expression of CTRP9 and improved neurobehavioral deficits and brain edema after ICH. Even though the endogenous expression of CTRP9 was increased after ICH, it may not be sufficient to significantly reduce the massive neuroinflammatory insult after ICH. Exogenous administration of rCTRP9 further potentiates potential protective pathways against neuroinflammation after ICH.
Previous studies report that CTRP9 increased insulin sensitivity and lowered blood sugar levels in the hyperglycemia model [22]. Thus, we tested blood glucose changes during the first 24 h after rCTRP9 administration. We observed that intranasal administration of rCTRP9 did not affect blood glucose levels. Blood glucose in all the groups, including vehicle or rCTRP9 groups, reached highest levels at 1 h post-surgery, possibly a response to surgery-related stress. Additionally, subsequent blood glucose levels continued to decline in all groups, reaching the lowest levels at 24 h after ICH. These findings can possibly be attributed to surgery-induced stress and hematoma that increases energy expenditure as well as reduce food intake capacity of the animals. Since there were no difference observed in blood glucose levels between groups after intranasal rCTRP9 administration, it is reasonable to consider that rCTRP9 did not cause hypoglycemia in our ICH model.
We found that AdipoR1 was expressed not only in microglia but also in neurons and astrocytes by immunofluorescence staining. These results are consistent with previous reports. Although adiponectin receptors have been shown to exist abundantly in the brain, its exact role in brain diseases remains unclear. Currently, two adiponectin receptors have been identified, AdipoR1 and AdipoR2. The expression of AdipoR1 has been reported to be more pronounced than AdipoR2 in the brain [35]. In addition, AdipoR1 and AdipoR2 have differential binding affinities to various adiponectin multimers. AdipoR1 binds to globular adiponectin (gAd) with high affinity, while AdipoR2 has an intermediate affinity with both gAd and full-length adiponectin [36]. In addition, AdipoR1 predominantly binds to adiponectin and suppresses NFκB activation and subsequently reduces the expression of pro-inflammatory cytokine, while AdipoR2 does not mediate the effects of adiponectin on microglial cells [6]. In this study, we observed that intracerebroventricular injection of AdipR1 siRNA decreased the expression of AdipoR1 and worsened neurological function in naïve and ICH mice. These results indicated a beneficial role of AdipR1 as well as demonstrated the knockdown efficacy AdipoR1.
The expression of AdipoR1 is regulated by many factors; obesity, alcohol, and insulin resistance can lower while exercise, stress, and cerebral ischemic conditions can increase the levels of AdipoR1. Our findings showed that AdipoR1 expression temporarily increased after ICH and reached peak levels at 24 h post-ICH. The receptor AdipoR1 is a high-affinity receptor for globular adiponectin. An increase in AdipoR1 expression may be necessary to perform its function given the increase in expression of globular domain of CTRP9 after ICH injury. This may be a possible explanation for the increase in AdipoR1 expression after ICH. Additionally, our results showed that AdipoR1 was modulated in a similar manner to CTRP9 levels after ICH, which indicates that rCTRP9 may exert its function through AdipoR1. Furthermore, rCTRP9 administration increased the expression of AdipoR1. The results were consistent with previous study which showed that adiponectin increased the expression of AdipoR1 and protected the brain against cerebral ischemic stroke [37]. We speculate that acute ICH injury may initially activate the inflammasome pathway and then trigger the stress reaction of adiponectin AdipoR1 signaling to reduce local inflammation. This is supported by the findings of our study which demonstrated that CTRP9/AdipoR1 showed anti-inflammatory effects in the ICH model, which is consistent with a previous study in which adiponectin was protective against cerebral ischemic stroke [38]. We measured inflammatory cytokines expression in the brain after ICH and with rCTRP9 administration. Local immune cells such as microglia and peripheral immune cells have been implicated in contributing to neuroinflammation after ICH. Since we administered rCTRP9 via intranasal administration which allows direct delivery of the agent to the brain, we measured changes in cytokine expression in the brain and levels of circulating cytokines were not measured in this study. Furthermore, to demonstrate the role of AdipoR1 in the anti-inflammatory mechanism of rCTRP9, AdipoR1 siRNA was administered by intracerebroventricular injection at 48 h before ICH with rCTRP9 treatment. The results showed that knockdown of AdipoR1 abolished the protective effects of rCTRP9 and reversed the expression of downstream proteins, suggesting that AdipoR1 played an essential role in the activating the downstream anti-inflammatory pathway by rCTRP9.
Neuroinflammation is a key process leading to secondary neurological damage after ICH [39]. Although anti-inflammatory effects of adiponectin have been previously reported in many disease models [40,41,42], the underlying mechanism remains unclear. Next, we investigated the downstream signaling pathway mediated via AdipoR1 activation. APPL1 is the only adiponectin receptor (AdipoR)-interacting protein that has been identified. The NH2-terminal intracellular region of AdipoR directly interacts with the PTB domain of APPL1. APPL1 was required for AdipoR-induced activation of downstream pathways to exert its anti-inflammatory and cytoprotective effects [43]. Our results showed that APPL1 expression increased with rCTRP9 administration after ICH. AMPK is a major downstream signaling molecule activated by APPL1 and has been demonstrated by studies on adiponectin-dependent AMPK signaling. A recent study suggested that bakkenolide B inhibited lipopolysaccharide-induced pro-inflammatory cytokines via AMPK activation in microglia [44]. In our study, administration of rCTRP9 markedly increased the expression of p-AMPK. This finding was consistent with a previous study in which CTRP9 protected against acute cardiac injury following ischemia-reperfusion via AMPK-dependent mechanism [45]. Due to its role in inducing the expression of proinflammatory cytokines, the nuclear factor NFκB pathway has long been considered as a typical proinflammatory signaling pathway. Prior published studies showed that the anti-inflammatory effects of adiponectin may be mediated via AMPK/NFκB signaling pathway [46]. In this study, we observed that rCTRP9 treatment upregulated the expression of AdipoR1, APPL1, and p-AMPK, and downregulated the pro-inflammatory p-NFκB after ICH. Furthermore, AdipoR1 siRNA and Dorsomorphin reversed the anti-inflammatory effects by rCTRP9. Overall, our results indicated that rCTRP9 treatment exerted anti-inflammatory effects through AdipoR1/AMPK/NFκB pathway in ICH model.
There are some limitations in the present study. First, AdipoR1 is expressed in astrocytes and may play a role in blood-brain barrier protective effects of rCTRP9, which may also contribute to the beneficial effects of rCTRP9 after ICH. Second, the pathophysiology of neuroinflammation after ICH is a complex network. Other downstream factors such as CAMKKβ and PPARα may be modulated by rCTRP9 administration after ICH which was not explored in this study. In the current study, we focused on APPL1/AMPK signaling anti-inflammatory mechanisms. Further studies need to be conducted to explore other potential signaling pathways contributed by rCTRP9 and AdipoR1 activation. Third, we did not evaluate the effects of CTRP9 in different age groups or in ICH with systemic co-morbidities. ICH tends to occur in older population with hypertension, vascular disorders, cerebral amyloid angiopathy, or anticoagulation. Previous publications indicate that adiponectin may have effect on hypertension and aging. Clinical studies have shown a relationship between serum adiponectin concentrations and the activity of renin-angiotensin-aldosterone system (RAAS), leading to changes in blood pressure [47, 48]. The effect of adiponectin on the cardiovascular system has been demonstrated partly by activation of the AMPK and cyclooxygenase-2 (COX-2) pathways, relaxation of vascular endothelial cells, reduction of endothelial cell apoptosis, and promotion of nitric oxide production [49, 50]. The lifespan of AdipoR1 or AdipoR2 knockout mice was reported to be shorter than that of control mice, the mechanisms involved included AMPK, mammalian target of rapamycin (mTOR) and SIRT, which are known to be key longevity molecules [51]. Overall, these findings indicate a need to study the relationship between adiponectin and CTRP9 with hypertension, aging, and systemic co-morbidities associated with ICH. Fourth, in this study, we did not evaluate sex-specific differences in the effects of CTRP9 in ICH. Estrogen has been reported to have neuroprotective effects, and it reduced neurological impairment after ICH and subarachnoid hemorrhage in rats [52, 53]. We therefore, used 8-week-old male adult CD1 mice for this study in order to eliminate the interference due to the effects of female sex hormones in the study outcomes.
Conclusions
In summary, we first showed that rCTRP9 administration improved neurological function, reduced brain edema, and alleviated inflammation through AdipoR1/AMPK/NFκB pathway after ICH. CTRP9, the closest analogue of adiponectin, may be a potential therapeutic target against neuroinflammation after ICH.
Abbreviations
- AdipoR1:
-
Adiponectin receptor 1
- AMPK:
-
Adenosine monophosphate-activated protein kinase
- APPL1:
-
Adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1
- BWC:
-
Brain water content
- CTRP9:
-
C1q/TNF-related protein 9
- DMSO:
-
Dimethyl sulfoxide
- GFAP:
-
Glial fibrillary acidic protein
- Iba-1:
-
Ionized calcium binding adaptor molecule-1
- ICH:
-
intracerebral hemorrhage
- IL-6:
-
Interleukin-6
- NeuN:
-
Neuronal nuclear
- p-AMPK:
-
Phosphorylated-AMPK
- PBS:
-
Phosphate-buffered saline
- p-NFκB:
-
Phosphorylated-nuclear factor kappa B
- Scr siRNA:
-
Scramble siRNA
- siRNA:
-
Small interfering ribonucleic acid
- TNFα:
-
Tumor necrosis factor
References
Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 2012;11:720–31.
Wilkinson DA, Pandey AS, Thompson BG, Keep RF, Hua Y, Xi G. Injury mechansims in acute intracerebral hemorrhage. Neuropharmacology. 2018;134:240–8.
Okamoto Y, Kihara S, Ouchi N, Nishida M, Arita Y, Kumada M, Ohashi K, Sakai N, Shimomura I, Kobayashi H, et al. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2002;106:2767–70.
Whitehead JP, Richards AA, Hickman IJ, Macdonald GA, Prins JB. Adiponectin--a key adipokine in the metabolic syndrome. Diabetes Obes Metab. 2006;8:264–80.
Kang KH, Higashino A, Kim HS, Lee YT, Kageyama T. Molecular cloning, gene expression, and tissue distribution of adiponectin and its receptors in the Japanese monkey, Macaca fuscata. J Med Primatol. 2009;38:77–85.
Nicolas S, Cazareth J, Zarif H, Guyon A, Heurteaux C, Chabry J, Petit-Paitel A. Globular adiponectin limits microglia pro-inflammatory phenotype through an AdipoR1/NF-kappaB signaling pathway. Front Cell Neurosci. 2017;11:352.
Bai S, Cheng L, Yang Y, Fan C, Zhao D, Qin Z, Feng X, Zhao L, Ma J, Wang X, et al. C1q/TNF-related protein 9 protects diabetic rat heart against ischemia reperfusion injury: role of endoplasmic reticulum stress. Oxidative Med Cell Longev. 2016;2016:1902025.
Sun Y, Yi W, Yuan Y, Lau WB, Yi D, Wang X, Wang Y, Su H, Wang X, Gao E, et al. C1q/tumor necrosis factor-related protein-9, a novel adipocyte-derived cytokine, attenuates adverse remodeling in the ischemic mouse heart via protein kinase A activation. Circulation. 2013;128:S113–20.
Myerburg MM, King JD Jr, Oyster NM, Fitch AC, Magill A, Baty CJ, Watkins SC, Kolls JK, Pilewski JM, Hallows KR. AMPK agonists ameliorate sodium and fluid transport and inflammation in cystic fibrosis airway epithelial cells. Am J Respir Cell Mol Biol. 2010;42:676–84.
Wang Y, Huang Y, Xu Y, Ruan W, Wang H, Zhang Y, Saavedra JM, Zhang L, Huang Z, Pang T. A dual AMPK/Nrf2 activator reduces brain inflammation after stroke by enhancing microglia M2 polarization. Antioxid Redox Signal. 2018;28:141–63.
Hattori Y, Suzuki K, Hattori S, Kasai K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension. 2006;47:1183–8.
Yang Z, Kahn BB, Shi H, Xue BZ. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem. 2010;285:19051–9.
Mancini SJ, White AD, Bijland S, Rutherford C, Graham D, Richter EA, Viollet B, Touyz RM, Palmer TM, Salt IP. Activation of AMP-activated protein kinase rapidly suppresses multiple pro-inflammatory pathways in adipocytes including IL-1 receptor-associated kinase-4 phosphorylation. Mol Cell Endocrinol. 2017;440:44–56.
Ma Q, Huang B, Khatibi N, Rolland W 2nd, Suzuki H, Zhang JH, Tang J. PDGFR-alpha inhibition preserves blood-brain barrier after intracerebral hemorrhage. Ann Neurol. 2011;70:920–31.
Zhang Y, Chen Y, Wu J, Manaenko A, Yang P, Tang J, Fu W, Zhang JH. Activation of dopamine D2 receptor suppresses Neuroinflammation through alphaB-crystalline by inhibition of NF-kappaB nuclear translocation in experimental ICH mice model. Stroke. 2015;46:2637–46.
Yu L, Lu Z, Burchell S, Nowrangi D, Manaenko A, Li X, Xu Y, Xu N, Tang J, Dai H, Zhang JH. Adropin preserves the blood-brain barrier through a Notch1/Hes1 pathway after intracerebral hemorrhage in mice. J Neurochem. 2017;143:750–60.
Krafft PR, Caner B, Klebe D, Rolland WB, Tang J, Zhang JH. PHA-543613 preserves blood-brain barrier integrity after intracerebral hemorrhage in mice. Stroke. 2013;44:1743–7.
Lekic T, Hartman R, Rojas H, Manaenko A, Chen W, Ayer R, Tang J, Zhang JH. Protective effect of melatonin upon neuropathology, striatal function, and memory ability after intracerebral hemorrhage in rats. J Neurotrauma. 2010;27:627–37.
Sherchan P, Lekic T, Suzuki H, Hasegawa Y, Rolland W, Duris K, Zhan Y, Tang J, Zhang JH. Minocycline improves functional outcomes, memory deficits, and histopathology after endovascular perforation-induced subarachnoid hemorrhage in rats. J Neurotrauma. 2011;28:2503–12.
Chen Y, Zhang Y, Tang J, Liu F, Hu Q, Luo C, Tang J, Feng H, Zhang JH. Norrin protected blood-brain barrier via frizzled-4/beta-catenin pathway after subarachnoid hemorrhage in rats. Stroke. 2015;46:529–36.
Ma K, Cabrero A, Saha PK, Kojima H, Li L, Chang BH, Paul A, Chan L. Increased beta-oxidation but no insulin resistance or glucose intolerance in mice lacking adiponectin. J Biol Chem. 2002;277:34658–61.
Wong GW, Krawczyk SA, Kitidis-Mitrokostas C, Ge G, Spooner E, Hug C, Gimeno R, Lodish HF. Identification and characterization of CTRP9, a novel secreted glycoprotein, from adipose tissue that reduces serum glucose in mice and forms heterotrimers with adiponectin. FASEB J. 2009;23:241–58.
Zheng Q, Yuan Y, Yi W, Lau WB, Wang Y, Wang X, Sun Y, Lopez BL, Christopher TA, Peterson JM, et al. C1q/TNF-related proteins, a family of novel adipokines, induce vascular relaxation through the adiponectin receptor-1/AMPK/eNOS/nitric oxide signaling pathway. Arterioscler Thromb Vasc Biol. 2011;31:2616–23.
Li J, Zhang P, Li T, Liu Y, Zhu Q, Chen T, Liu T, Huang C, Zhang J, Zhang Y, Guo Y. CTRP9 enhances carotid plaque stability by reducing pro-inflammatory cytokines in macrophages. Biochem Biophys Res Commun. 2015;458:890–5.
Kambara T, Shibata R, Ohashi K, Matsuo K, Hiramatsu-Ito M, Enomoto T, Yuasa D, Ito M, Hayakawa S, Ogawa H, et al. C1q/tumor necrosis factor-related protein 9 protects against acute myocardial injury through an adiponectin receptor I-AMPK-dependent mechanism. Mol Cell Biol. 2015;35:2173–85.
Wu CH, Shyue SK, Hung TH, Wen S, Lin CC, Chang CF, Chen SF. Genetic deletion or pharmacological inhibition of soluble epoxide hydrolase reduces brain damage and attenuates neuroinflammation after intracerebral hemorrhage. J Neuroinflammation. 2017;14:230.
Yang Z, Zhao T, Zou Y, Zhang JH, Feng H. Curcumin inhibits microglia inflammation and confers neuroprotection in intracerebral hemorrhage. Immunol Lett. 2014;160:89–95.
Ma B, Day JP, Phillips H, Slootsky B, Tolosano E, Dore S. Deletion of the hemopexin or heme oxygenase-2 gene aggravates brain injury following stroma-free hemoglobin-induced intracerebral hemorrhage. J Neuroinflammation. 2016;13:26.
Toussirot E, Streit G, Nguyen NU, Dumoulin G, Le Huede G, Saas P, Wendling D. Adipose tissue, serum adipokines, and ghrelin in patients with ankylosing spondylitis. Metabolism. 2007;56:1383–9.
Brochu-Gaudreau K, Rehfeldt C, Blouin R, Bordignon V, Murphy BD, Palin MF. Adiponectin action from head to toe. Endocrine. 2010;37:11–32.
Yang Q, Fu C, Xiao J, Ye Z. Uric acid upregulates the adiponectinadiponectin receptor 1 pathway in renal proximal tubule epithelial cells. Mol Med Rep. 2018;17:3545–54.
Delaigle AM, Jonas JC, Bauche IB, Cornu O, Brichard SM. Induction of adiponectin in skeletal muscle by inflammatory cytokines: in vivo and in vitro studies. Endocrinology. 2004;145:5589–97.
Shen L, Miao J, Yuan F, Zhao Y, Tang Y, Wang Y, Zhao Y, Yang GY. Overexpression of adiponectin promotes focal angiogenesis in the mouse brain following middle cerebral artery occlusion. Gene Ther. 2013;20:93–101.
Huang H, Iida KT, Sone H, Ajisaka R. The regulation of adiponectin receptors expression by acute exercise in mice. Exp Clin Endocrinol Diabetes. 2007;115:417–22.
Guillod-Maximin E, Roy AF, Vacher CM, Aubourg A, Bailleux V, Lorsignol A, Penicaud L, Parquet M, Taouis M. Adiponectin receptors are expressed in hypothalamus and colocalized with proopiomelanocortin and neuropeptide Y in rodent arcuate neurons. J Endocrinol. 2009;200:93–105.
Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–9.
Guo F, Jiang T, Song W, Wei H, Wang F, Liu L, Ma L, Yin H, Wang Q, Xiong L. Electroacupuncture attenuates cerebral ischemia-reperfusion injury in diabetic mice through adiponectin receptor 1-mediated phosphorylation of GSK-3beta. Mol Neurobiol. 2015;51:685–95.
Nishimura M, Izumiya Y, Higuchi A, Shibata R, Qiu J, Kudo C, Shin HK, Moskowitz MA, Ouchi N. Adiponectin prevents cerebral ischemic injury through endothelial nitric oxide synthase dependent mechanisms. Circulation. 2008;117:216–23.
Wang J, Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27:894–908.
Ohashi K, Shibata R, Murohara T, Ouchi N. Role of anti-inflammatory adipokines in obesity-related diseases. Trends Endocrinol Metab. 2014;25:348–55.
Yang Y, Hu W, Jiang S, Wang B, Li Y, Fan C, Di S, Ma Z, Lau WB, Qu Y. The emerging role of adiponectin in cerebrovascular and neurodegenerative diseases. Biochim Biophys Acta. 2015;1852:1887–94.
Wang Y, Zhou M, Lam KS, Xu A. Protective roles of adiponectin in obesity-related fatty liver diseases: mechanisms and therapeutic implications. Arq Bras Endocrinol Metabol. 2009;53:201–12.
Deepa SS, Dong LQ. APPL1: role in adiponectin signaling and beyond. Am J Physiol Endocrinol Metab. 2009;296:E22–36.
Park SY, Choi MH, Park G, Choi YW. Petasites japonicus bakkenolide B inhibits lipopolysaccharideinduced proinflammatory cytokines via AMPK/Nrf2 induction in microglia. Int J Mol Med. 2018;41:1683–92.
Kambara T, Ohashi K, Shibata R, Ogura Y, Maruyama S, Enomoto T, Uemura Y, Shimizu Y, Yuasa D, Matsuo K, et al. CTRP9 protein protects against myocardial injury following ischemia-reperfusion through AMP-activated protein kinase (AMPK)-dependent mechanism. J Biol Chem. 2012;287:18965–73.
Devaraj S, Torok N, Dasu MR, Samols D, Jialal I. Adiponectin decreases C-reactive protein synthesis and secretion from endothelial cells: evidence for an adipose tissue-vascular loop. Arterioscler Thromb Vasc Biol. 2008;28:1368–74.
Yilmaz MI, Sonmez A, Kilic S, Celik T, Bingol N, Pinar M, Mumcuoglu T, Ozata M. The association of plasma adiponectin levels with hypertensive retinopathy. Eur J Endocrinol. 2005;152:233–40.
Nowak L, Adamczak M, Wiecek A. Blockade of sympathetic nervous system activity by rilmenidine increases plasma adiponectin concentration in patients with essential hypertension. Am J Hypertens. 2005;18:1470–5.
Wang ZV, Scherer PE. Adiponectin, cardiovascular function, and hypertension. Hypertension. 2008;51:8–14.
Ouedraogo R, Gong Y, Berzins B, Wu X, Mahadev K, Hough K, Chan L, Goldstein BJ, Scalia R. Adiponectin deficiency increases leukocyte-endothelium interactions via upregulation of endothelial cell adhesion molecules in vivo. J Clin Invest. 2007;117:1718–26.
Okada-Iwabu M, Yamauchi T, Iwabu M, Honma T, Hamagami K, Matsuda K, Yamaguchi M, Tanabe H, Kimura-Someya T, Shirouzu M, et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature. 2013;503:493–9.
Nakamura T, Hua Y, Keep RF, Park JW, Xi G, Hoff JT. Estrogen therapy for experimental intracerebral hemorrhage in rats. J Neurosurg. 2005;103:97–103.
Shih HC, Lin CL, Lee TY, Lee WS, Hsu C. 17beta-estradiol inhibits subarachnoid hemorrhage-induced inducible nitric oxide synthase gene expression by interfering with the nuclear factor kappa B transactivation. Stroke. 2006;37:3025–31.
Funding
This study is supported partially by grants from National Institutes of Health (NS091042 and NS082184) to Dr. J. H. Zhang.
Availability of data and materials
The data, analytic methods, and study materials will be made available to other researchers for the purpose of reproducing the results or replicating the procedures. The data that support the findings of this study are available from the corresponding author upon reasonable request. Authors will be responsible for maintaining availability.
Author information
Authors and Affiliations
Contributions
JHZ, LHZ, and PS conceived and designed the study. LHZ, SPC, and JPT conducted the experiments, analyzed the data, and drafted the manuscript. PS and YD worked on the manuscript revision. SPC and JPT participated in the experimental design, data analysis and interpretation, and manuscript preparation. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
All animal experiments were approved by the Institutional Animal Care and Use Committee at Loma Linda University. The study followed the Health’s Guide for the Care and Use of Laboratory Animals (National Research Council) and complied with the ARRIVE guidelines for reporting in vivo experiments.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Experimental design and animal groups. ICH, intracerebral hemorrhage; rCTRP9, recombinant C1q/TNF-related protein 9; WB, western blot; IHC, immunohistochemistry; BS, blood sugar; siRNA, small interfering ribonucleic acid. (TIF 2484 kb)
Additional file 2:
Table S1. Summary of experimental groups and mortality rate in the study. (DOCX 21 kb)
Additional file 3:
Figure S2. Schematic mechanism of the effects of rCTRP9 on anti-neuroinflammation after ICH. rCTRP9, recombinant C1q/TNF-related protein 9; AdipoR1, adiponectin receptor 1; APPL1, adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1; p-AMPK, phosphorylated-adenosine monophosphate-activated protein kinase; p-NFκB, phosphorylated-nuclear factor kappa B; TNFα, tumor necrosis factor α; IL-6, interleukin-6. (TIF 387 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhao, L., Chen, S., Sherchan, P. et al. Recombinant CTRP9 administration attenuates neuroinflammation via activating adiponectin receptor 1 after intracerebral hemorrhage in mice. J Neuroinflammation 15, 215 (2018). https://doi.org/10.1186/s12974-018-1256-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-018-1256-8