
Abstract
Background
The incidence of stage I and stage II lung adenocarcinoma (LUAD) is likely to increase with the introduction of annual screening programs for high-risk individuals. We aimed to identify a reliable prognostic signature with immune-related genes that can predict prognosis and help making individualized management for patients with early-stage LUAD.
Methods
The public LUAD cohorts were obtained from the large-scale databases including 4 microarray data sets from the Gene Expression Omnibus (GEO) and 1 RNA-seq data set from The Cancer Genome Atlas (TCGA) LUAD cohort. Only early-stage patients with clinical information were included. Cox proportional hazards regression model was performed to identify the candidate prognostic genes in GSE30219, GSE31210 and GSE50081 (training set). The prognostic signature was developed using the overlapped prognostic genes based on a risk score method. Kaplan–Meier curve with log-rank test and time-dependent receiver operating characteristic (ROC) curve were used to evaluate the prognostic value and performance of this signature, respectively. Furthermore, the robustness of this prognostic signature was further validated in TCGA-LUAD and GSE72094 cohorts.
Results
A prognostic immune signature consisting of 21 immune-related genes was constructed using the training set. The prognostic signature significantly stratified patients into high- and low-risk groups in terms of overall survival (OS) in training data set, including GSE30219 (HR = 4.31, 95% CI 2.29–8.11; P = 6.16E−06), GSE31210 (HR = 11.91, 95% CI 4.15–34.19; P = 4.10E−06), GSE50081 (HR = 3.63, 95% CI 1.90–6.95; P = 9.95E−05), the combined data set (HR = 3.15, 95% CI 1.98–5.02; P = 1.26E−06) and the validation data set, including TCGA-LUAD (HR = 2.16, 95% CI 1.49–3.13; P = 4.54E−05) and GSE72094 (HR = 2.95, 95% CI 1.86–4.70; P = 4.79E−06). Multivariate cox regression analysis demonstrated that the 21-gene signature could serve as an independent prognostic factor for OS after adjusting for other clinical factors. ROC curves revealed that the immune signature achieved good performance in predicting OS for early-stage LUAD. Several biological processes, including regulation of immune effector process, were enriched in the immune signature. Moreover, the combination of the signature with tumor stage showed more precise classification for prognosis prediction and treatment design.
Conclusions
Our study proposed a robust immune-related prognostic signature for estimating overall survival in early-stage LUAD, which may be contributed to make more accurate survival risk stratification and individualized clinical management for patients with early-stage LUAD.
Similar content being viewed by others


Background
Lung cancer is the leading cause of death from cancer. In the United States, there will be approximately 228,820 newly diagnosed cases and 135,720 deaths in 2020 [1]. Lung adenocarcinoma (LUAD) is the most common histological type and accounts for nearly 60% of non-small cell lung cancer (NSCLC), which comprises approximately 85% of lung cancer [2,3,4]. Surgical lobectomy remains the preferred treatment strategy for patients with operable early-stage LUAD [5]. Although patients with early-stage LUAD have a relatively superior prognosis, nearly 10–44% of these patients still die within 5 years after surgical intervention [6, 7]. Recently, several studies revealed that adjuvant chemotherapy brought a clear 5-year survival benefit ranged from 4 to 10% for patients with resected stage II LUAD and can be considered for stage IB LUAD with primary tumor more than 4 cm [8,9,10,11], but not for patients with stage IA because of the potential detrimental effect [12]. Thus, besides the traditional clinical factors, it is imperative to develop a novel prognostic signature to perform personalized survival risk stratification and identify the high-risk early-stage patients who might benefit from additional systemic therapy.
In recent years, numerous studies have reported prognostic signatures to make survival stratification and predict prognosis for patients with LUAD using genomics and transcriptomics data [13,14,15]. Unfortunately, the signatures proposed by these studies have not been incorporated into clinical practice owing to the problems such as small sample size and insufficient independent validation [16, 17]. Nowadays, the available public, large-scale databases containing enough gene expression data, such as TCGA (The Cancer Genome Atlas) and GEO (Gene Expression Omnibus) database, bring the opportunity to make more reliable prognostic signatures for lung cancer [18]. Immune system has been shown to play a crucial role in cancer initiation and progression [19, 20]. In addition, avoiding immune destruction has been accepted as a novel hallmark of cancer [21]. Recently, immunotherapies have achieved a notably and durable response in LUAD by targeting specific immune checkpoints like PD-1 or PD-L1 [22, 23]. Several studies have reported immune-related gene signatures which could predict prognosis and provide potential targets for immunotherapy in patients with LUAD [24,25,26]. However, few prognostic models have focused on immune-related genes in early-stage LUAD.
In this study, we used the gene expression data sets from GEO and TCGA to develop and validate a prognostic prediction model for early-stage LUAD based on immune-related genes. A novel 21-gene based prognostic immune signature with robust prediction power for early-stage LUAD was developed, which allows clinicians to evaluate the prognosis of patients with early-stage LUAD and might provide promise for individualized therapeutic interventions.
Methods
Data preprocessing
We downloaded four independent NSCLC microarray data sets from GEO database (https://www.ncbi.nlm.nih.gov/geo/) using the GEOquery package [27]. Only early-stage LUAD patients were included. Patients without survival status or whose overall survival time shorter than 30 days were removed from the study. Among these data sets, the gene expression data of GSE30219 [28], GSE31210 [29, 30] and GES50081 [31] were generated by the same platform GPL570 (Affymetrix Human Genome U133 Plus 2.0 Array). These data sets were defined as training set and selected to screen for the candidate prognostic genes, while GSE72094 [32], another microarray data set, was chose for independent validation. Besides, The gene expression data and corresponding clinical information of TCGA-LUAD cohort, a RNA-seq data set, were downloaded by the UCSC Xena platform [33, 34], which was used for another independent validation. The general information of these datasets was summarized in Additional file 1: Table S1. The gene expression data of GEO and TCGA-LUAD data sets were normalized by the limma and DESeq2 package, respectively [35, 36]. Overall, a total of 1091 patients were enrolled in our study, including 82 patients from GSE30219, 204 patients from GSE31210, 127 patients from GSE50081, 311 patients from GSE72094 and 367 patients from TCGA-LUAD. The baseline characteristics of the patients enrolled in the study were described in Additional file 2: Table S2.
Development of the prognostic gene signature
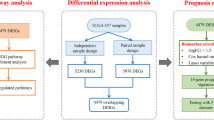
We constructed the prognostic gene signature by focusing on the immune-related genes, which were downloaded from the InnateDB database (https://innatedb.com/) [37]. The list of the immune-related genes was summarized in Additional file 3: Table S3. The flow chart of this study was presented in Fig. 1. Firstly, univariate cox proportional hazards regression model was performed to screen for the candidate prognostic genes (p < 0.05) associated with OS in GSE30219, GSE31210 and GSE50081 cohort, respectively. Candidate genes with Hazard ratio (HR) > 1 were considered as risky prognostic genes, while HR < 1 as protective prognostic genes. The overlapped candidate prognostic genes were selected to develop the prognostic signature based on risk score model. In addition, the three microarray data sets were merged into 1 combined data set for further analysis.

Flow chart of this study
Then a risk score for each patient was established based on a linear combination of the overlapped candidate prognostic genes expression levels weighted by the regression coefficient (β) derived from the univariate cox regression analysis [38, 39]. The risk score formula was defined as the following:
The n, expi and βi in the above formula represent the number of prognostic genes, the expression value and the coefficient of gene i, respectively [40]. Optimal cutoff value of the risk score in each data set was determined by the survminer package in R [41]. According to the cutoff value, patients were classified into high- and low-risk groups.
Evaluation of the immune-related prognostic signature
To assess the prognostic value of this prognostic signature, we firstly estimated the survival curves between the high- and low-risk groups by the Kaplan–Meier method using the survival and survminer package in GSE30219, GSE31210, GSE50081 and the combined data set, respectively [41, 42], with log-rank test to determine the statistical significances in OS between two groups. Meanwhile, time-dependent receiver operating characteristic (ROC) curve was conducted to evaluate the performance of this signature by calculating the area under the ROC curves (AUC) using timeROC package [43]. Furthermore, the same risk score formula was employed on GSE72094 and TCGA-LUAD cohort, which were served as independent validation data sets, to further evaluate and validate the efficiency of this signature.
Functional annotation and enrichment analysis
To acquire the potential biological processes of the overlapped prognostic genes, Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were performed using clusterProfiler package [44].
Statistical analysis
All statistical analyses were performed using R (version 3.6.2; R Foundation for Statistical Computing) [45] and RStudio (version 1.2.1335) (https://rstudio.com/). To investigate whether the gene signature was an independent prognostic factor for early-stage LUAD, univariate analysis was performed to evaluate the association of the gene signature and other clinical parameters with overall survival. Risk factors (p < 0.2) derived from univariate analysis were selected for further analysis in multivariate cox regression model [46, 47]. Heatmap was generated using the pheatmap package [48]. The detailed information of the system, software and packages using in the study were summarized in Additional file 4: Table S4. p < 0.05 deemed statistically significant.
Results
Identification of 21 immune-related prognostic genes in the training set
A total of 1091 patients with early-stage LUAD (533 men [49%], 558 women [51%]; median age [range], 66 [30–89] years), including 413 patients in the training set and 678 patients in the validation set, were enrolled in the analysis. Among 1051 immune-related genes from the innateDB database, 920 genes were measured in the training set. Under the cutoff value of p < 0.05, 173 genes in GSE30219, 300 genes in GSE31210 and 146 genes in GSE50081 were identified as candidate prognostic genes which were significantly associated with OS. After overlapping these prognostic genes among these data sets, 21 overlapped genes were finally screened, including 14 risky genes and 7 protective genes. The general information of the overlapped genes and corresponding coefficients were summarized in Additional file 5: Table S5.
Development of the 21-gene based immune-related prognostic signature
We calculated the 21-gene based risk score for each patient in the training set using the risk score formula (Additional file 6: Table S6). Patients were classified into high- and low-risk groups using the optimal cutoff analyzed by the survminer package. The cutoff value in each cohort was summarized in Additional file 7: Table S7. The distribution of the risk scores, survival status and the expression levels of the 21 genes in the training set were shown in Additional file 8: Figure. S1.
Kaplan–Meier survival curves revealed that patients in the high-risk group shown significantly poorer OS than patients in the low-risk group (Fig. 2a). Moreover, the AUCs for 1-year, 3-year and 5-year were 0.75, 0.80 and 0.82 in GSE30219, 0.78, 0.75 and 0.81 in GSE31210 and 0.73, 0.75 and 0.74 in GSE50081, respectively (Fig. 2b), suggesting that this 21-gene signature achieved a relatively high performance for early-stage LUAD survival prediction. Furthermore, we conducted survival analysis in the combined data set to assess the reliability of this signature. Consistent with the results of single data set in the training set, Kaplan–Meier curve showed that patients in the high-risk group exhibited shorter OS than those in the low-risk group (p < 0.0001) (Fig. 2a). The AUCs for 1-year, 3-year and 5-year were 0.66, 0.66 and 0.70, respectively (Fig. 2b), implying that this gene signature also had a good performance for prognosis prediction in the combined data set.

Correlation between the 21-gene signature and Overall survival in the training set (early-stage LUAD). a Kaplan–Meier survival curves between high- and low-risk groups, b ROC curves for 1-year, 3-year and 5-year survival prediction by the 21-gene signature
External validation of the 21-gene prognostic signature
To further validate the robustness of the 21-gene signature, the risk score for each patient was calculated using the same risk score formula in two independent data sets, including TCGA-LUAD and GSE72094 cohort. We divided the patients into high- and low-risk group according to the optimal cutoff. Consistent with the results in the training set, patients of high-risk group shown conspicuously poorer OS than those of low-risk group in both TCGA-LUAD and GSE72094 cohort (p < 0.0001, Fig. 3a). The AUCs for 1-year, 3-year and 5-year were 0.61, 0.66 and 0.62 in TCGA-LUAD, and 0.70, 0.64 and 0.94 in GSE72094 (Fig. 3b), which implies that the prognostic signature has a valid performance for OS prediction in validation data sets. The distribution of the risk scores, survival status and the expression levels of the 21 genes in the validation set were shown in Additional file 8: Figure. S1. The data for Kaplan–Meier survival analysis and ROC analysis were summarized in Additional file 9: Table S8. Taken together, these results suggest that this 21-gene based prognostic signature is robust in prognosis prediction for early-stage LUAD and can be used in both microarray and RNA-sequencing data sets.

Correlation between the 21-gene signature and Overall survival in the validation set (early-stage LUAD). a Kaplan–Meier survival curves between high- and low-risk groups in TCGA-LUAD and GSE72094 cohort, respectively. b ROC curves for 1-year, 3-year and 5-year survival prediction by the 21-gene signature in TCGA-LUAD and GSE72094 cohort, respectively
The 21-gene prognostic signature is an independent prognostic factor
Univariate and multivariate cox analysis were performed in both training and validation sets to investigate whether this 21-gene prognostic signature could be served as an independent prognostic factor for patients with early-stage LUAD. The prognostic signature and other available clinicopathological factors were included for analysis. Univariate regression analysis indicated that the prognostic signature was significantly associated with OS for early-stage LUAD in GSE30219 (HR = 4.31, 95% CI 2.29–8.11, P = 6.16E−06), GSE31210 (HR = 11.91, 95% CI 4.15–34.19; P = 4.10E−06), GSE50081 (HR = 3.63, 95% CI 1.90–6.95; P = 9.95E−05), combined data set (HR = 3.15, 95% CI 1.98–5.02; P = 1.26E−06) (Table 1), TCGA-LUAD (HR = 2.16, 95% CI 1.49–3.13; P = 4.54E−05) and GSE72094 (HR = 2.95, 95% CI 1.86–4.70; P = 4.79E−06) (Table 2). Then, risk factors (P < 0.2) derived from the univariate analysis were selected for further multivariate analysis. The results shown that there was a significantly association between the prognostic signature and OS in GSE30219 (HR = 5.01, 95% CI 2.50–10.06; P = 5.75E−06), GSE31210 (HR = 8.82, 95% CI 2.86–27.14; P = 1.48E−04), GSE50081 (HR = 2.74, 95% CI 1.37–5.46; P = 4.24E−03), combined data set (HR = 3.01, 95% CI 1.86–4.85; P = 6.44E−06) (Table 1), TCGA-LUAD (HR = 1.91, 95% CI 1.28–2.85; P = 1.55E−03) and GSE72094 (HR = 2.94, 95% CI 1.81–4.79; 1.47E−05) (Table 2). These results demonstrated that the 21-gene based prognostic signature was an independent prognostic factor for patients with early-stage LUAD in both training set and validation set after adjusting for other clinical and pathologic factors.
Prognosis prediction by combining the 21-gene prognostic signature with stage
Multivariate analysis revealed that the prognostic signature and stage were both independent prognostic factors in the combined data set, suggesting a complementary value. Therefore, we attempted to develop an integrated prognostic model for survival prediction by combining the prognostic signature with tumor stage in the combined data set. Based on the risk and stage, patients were classified into six groups: group 1 (stage IA with low-risk), group 2 (stage IA with high-risk), group 3 (stage IB with low-risk), group 4 (stage IB with high-risk), group 5 (stage II with low-risk) and group 6 (stage II with high-risk) (Fig. 4). Kaplan–Meier survival analysis were performed between different groups. The results revealed that patients in group 2, group3, group 4, group5 and group 6 had worse prognosis compared with patients in group 1, with group 1 exhibited the best prognosis and group 6 showed the worst (Fig. 4). Nevertheless, there was no significant difference between patients in group 2 and group 3/4/5 (Fig. 4). These results display that patients of stage IA with high-risk have similar prognosis to those of stage IB and stage II with low-risk, suggesting adjuvant chemotherapy might be beneficial for stage IA LUAD with high-risk. Additionally, patients of early-stage LUAD could be divided into six different groups based on the stage and prognostic signature, which might be a more precise scheme to predict prognosis for patients with early-stage LUAD in the future practice.

Kaplan–Meier curves of overall survival for patients grouped by stage and 21-gene signature combination (early-stage LUAD)
Functional annotation and enrichment analysis of the 21-gene prognostic signature
To identify the underlying biological processes and pathways within this 21-gene signature, we performed GO enrichment and KEGG pathway analysis. The results indicated these genes were mainly enriched in biological processes such as positive regulation of cytokine production (GO:0001819), regulation of immune effector process (GO:0002697) and intrinsic apoptotic signaling pathway (GO:0097193) (Fig. 5). In addition, KEGG analysis revealed that several pathways like viral carcinogenesis, proteoglycans in cancer and Fc-gamma R-mediated phagocytosis (Fig. 5) were enriched among these genes.

Functional enrichment analysis of the 21 prognostic genes. a Gene Ontology analysis, b Kyoto Encyclopedia of Genes and Genomes pathway analysis
Discussion
Previous studies have reported different prognostic biomarkers for patients with early-stage LUAD [28, 49,50,51]. However, none of these studies focused on the immune-related genes in prognosis prediction. Recently, several studies have proposed prognostic signatures using immune-related genes for LUAD [24,25,26]. Nevertheless, some concerns hamper the prediction power of these prognostic signatures, such as insufficient sample size, lack of external independent validation or effective validation. In the present study, we developed a novel prognostic signature based on 21 immune-related genes for early-stage LUAD and validated it in two independent cohorts. Our prognostic signature was significantly associated with OS for early-stage LUAD and could further identify the high- and low-risk early-stage LUAD patients with significant differences in OS. Besides, the 21-gene prognostic signature showed a good prediction performance in all enrolled studies including GEO and TCGA-LUAD data sets, suggesting that our signature had a cross-platform compatibility. Multivariate regression analysis revealed that the 21-gene prognostic signature was an independent prognostic factor for all enrolled studies. These results suggest that the 21-gene prognostic signature could effectively predict the overall survival for early-stage LUAD.
In the aera of immunotherapy, it may hold great promise to discover prognostic and predictive biomarkers that are related to tumor immune microenvironment, which can be used for identifying novel molecular targets for patients [52]. Our functional enrichment analysis suggests that the genes in our prognostic signature are widely involved in the immune process. Among all the 21 prognostic genes, 14 (e.g., AQP3, BIRC5, C5AR1, HMOX1, IL32, IL6ST, MIF, MMP12, PLAUR, PMAIP1, RAC1, SMAD6, SPHK1, USP7) have been reported to be served as a prognostic biomarker or suggested to be a novel therapeutic targets for lung adenocarcinoma [53,54,55,56,57,58,59,60,61,62,63,64,65,66,67]. The remaining 7 genes, including ARF6, C7, ELF4, ITPR1, MOV10, PTCH1 and RIPK2, have not been previously reported to be associated with LUAD prognosis and might act as potential biomarker. We were particularly interested in studying ARF6, which was a member of small GTPases ADP-ribosylation factor family, and its downstream effector AMAP1 have been reported overexpressed in several types of cancer and could promote cancer cell proliferation, invasion and migration [68,69,70,71]. For example, KRAS and TP53 oncogenes could promote PD-L1 recycling and cell surface expression through ARF6-AMAP1 pathway, which is significantly involved in the immune evasion of pancreatic ductal adenocarcinoma cells [72].
Currently, tumor staging system has been widely used for prognosis prediction and treatment design for LUAD. However, prognosis might vary in patients with same stage owing to the variabilities in clinical behavior caused by genomic changes [73, 74]. Thus, it is critically needed to develop reliable prognostic biomarkers to predict prognosis and help clinical oncologists optimally select early-stage patients who might obtain survival benefit from additional system therapy. In the integrated prediction model analysis, early-stage LUAD patients could be stratified into six different groups by combining our 21-gene prognostic signature with tumor stage. Besides, no statistical significance exists in prognosis between stage IA patients with high-risk and stage II patients with low-risk. These findings may help clinicians identify high-risk patients and make individualized treatment design for these patients.
The limitations in our study need to be noted. First, although different cohorts from GEO and TCGA databases have been included in our study to develop and validate the immune-related prognostic signature, the study presents a retrospective design. Future large-scale prospective clinical studies needed to confirm our findings. Second, the data of specific mutations such as EGFR, KRAS and TP53 were only available in GSE31210 and GSE72094 cohort, thus it might be insufficient to assess the 21-gene prognostic signature with the specific mutations. Finally, the biological mechanisms of these prognostic genes in early-stage LUAD and the association of the prognostic signature with several prognostic biomarker such as PD-L1, IL-7R [75], CD8+ [76], are still unknown, Future studies are required to explore and clarify molecular functions of these immune-related prognostic genes during early-stage LUAD progression and the association between these genes with above prognostic biomarkers.
Conclusions
In summary, we developed and validated a promising immune-related prognostic signature comprising of 21 immune-related genes, which could serve as an independent prognostic biomarker for OS prediction in early-stage LUAD. Furthermore, a prediction model by combining our prognostic signature with tumor stage could more accurately evaluate patient’s prognosis. These findings might provide novel therapeutic targets and be used for making individualized management and hold promise for improving survival for patients with early-stage LUAD.
Availability of data and materials
The data sets analyzed in this study are available on the public databases.
Abbreviations
- LUAD:
-
Lung adenocarcinoma
- GEO:
-
Gene Expression Omnibus
- TCGA:
-
The Cancer Genome Atlas
- ROC:
-
Receiver operating characteristic
- OS:
-
Overall survival
- NSCLC:
-
Non-small cell lung cancer
- HR:
-
Hazard ratio
- AUC:
-
Area under the curve
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
Behera M, Owonikoko TK, Gal AA, Steuer CE, Kim S, Pillai RN, Khuri FR, Ramalingam SS, Sica GL. Lung adenocarcinoma staging using the 2011 IASLC/ATS/ERS classification: a pooled analysis of adenocarcinoma in situ and minimally invasive adenocarcinoma. Clin Lung Cancer. 2016;17:e57–e64.
Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–50.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29.
Neal RD, Sun F, Emery JD, Callister ME. Lung cancer. BMJ. 2019;365:l1725.
Goldstraw P, Chansky K, Crowley J, Rami-Porta R, Asamura H, Eberhardt WE, Nicholson AG, Groome P, Mitchell A, Bolejack V, et al. The IASLC lung cancer staging project: proposals for revision of the TNM stage groupings in the forthcoming (Eighth) edition of the TNM classification for lung cancer. J Thorac Oncol. 2016;11:39–51.
Rami-Porta R, Bolejack V, Crowley J, Ball D, Kim J, Lyons G, Rice T, Suzuki K, Thomas CF Jr, Travis WD, et al. The IASLC lung cancer staging project: proposals for the revisions of the T descriptors in the forthcoming eighth edition of the TNM classification for lung cancer. J Thorac Oncol. 2015;10:990–1003.
Burdett S, Pignon JP, Tierney J, Tribodet H, Stewart L, Le Pechoux C, Auperin A, Le Chevalier T, Stephens RJ, Arriagada R, et al. Adjuvant chemotherapy for resected early-stage non-small cell lung cancer. Cochrane Database Syst Rev. 2015. https://doi.org/10.1002/14651858.CD011430.
Liang Y, Wakelee HA. Adjuvant chemotherapy of completely resected early stage non-small cell lung cancer (NSCLC). Transl Lung Cancer Res. 2013;2:403–10.
McDonald F, De Waele M, Hendriks LE, Faivre-Finn C, Dingemans AC, Van Schil PE. Management of stage I and II nonsmall cell lung cancer. Eur Respir J. 2017. https://doi.org/10.1183/13993003.00764-2016.
Strauss GM, Herndon JE 2nd, Maddaus MA, Johnstone DW, Johnson EA, Harpole DH, Gillenwater HH, Watson DM, Sugarbaker DJ, Schilsky RL, et al. Adjuvant paclitaxel plus carboplatin compared with observation in stage IB non-small-cell lung cancer: CALGB 9633 with the Cancer and Leukemia Group B, Radiation Therapy Oncology Group, and North Central Cancer Treatment Group Study Groups. J Clin Oncol. 2008;26:5043–51.
Pignon JP, Tribodet H, Scagliotti GV, Douillard JY, Shepherd FA, Stephens RJ, Dunant A, Torri V, Rosell R, Seymour L, et al. Lung adjuvant cisplatin evaluation: a pooled analysis by the LACE Collaborative Group. J Clin Oncol. 2008;26:3552–9.
Director's Challenge Consortium for the Molecular Classification of Lung A, Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, Gerald WL, Eschrich S, Jurisica I, Giordano TJ, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med. 2008;14:822–7.
Li X, Shi Y, Yin Z, Xue X, Zhou B. An eight-miRNA signature as a potential biomarker for predicting survival in lung adenocarcinoma. J Transl Med. 2014;12:159.
Shi X, Tan H, Le X, Xian H, Li X, Huang K, Luo VY, Liu Y, Wu Z, Mo H, et al. An expression signature model to predict lung adenocarcinoma-specific survival. Cancer Manag Res. 2018;10:3717–32.
Subramanian J, Simon R. Gene expression-based prognostic signatures in lung cancer: ready for clinical use? J Natl Cancer Inst. 2010;102:464–74.
Tang H, Wang S, Xiao G, Schiller J, Papadimitrakopoulou V, Minna J, Wistuba II, Xie Y. Comprehensive evaluation of published gene expression prognostic signatures for biomarker-based lung cancer clinical studies. Ann Oncol. 2017;28:733–40.
Li B, Cui Y, Diehn M, Li R. Development and validation of an individualized immune prognostic signature in early-stage nonsquamous non-small cell lung cancer. JAMA Oncol. 2017;3:1529–37.
Angell H, Galon J. From the immune contexture to the Immunoscore: the role of prognostic and predictive immune markers in cancer. Curr Opin Immunol. 2013;25:261–7.
Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938–45.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Hellmann MD, Rizvi NA, Goldman JW, Gettinger SN, Borghaei H, Brahmer JR, Ready NE, Gerber DE, Chow LQ, Juergens RA, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017;18:31–41.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–544.
Luo C, Lei M, Zhang Y, Zhang Q, Li L, Lian J, Liu S, Wang L, Pi G, Zhang Y. Systematic construction and validation of an immune prognostic model for lung adenocarcinoma. J Cell Mol Med. 2020;24:1233–44.
Song Q, Shang J, Yang Z, Zhang L, Zhang C, Chen J, Wu X. Identification of an immune signature predicting prognosis risk of patients in lung adenocarcinoma. J Transl Med. 2019;17:70.
Zhang M, Zhu K, Pu H, Wang Z, Zhao H, Zhang J, Wang Y. An immune-related signature predicts survival in patients with lung adenocarcinoma. Front Oncol. 2019;9:1314.
Davis S, Meltzer PS. GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics. 2007;23:1846–7.
Rousseaux S, Debernardi A, Jacquiau B, Vitte AL, Vesin A, Nagy-Mignotte H, Moro-Sibilot D, Brichon PY, Lantuejoul S, Hainaut P, et al. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci Transl Med. 2013;5:186ra166.
Okayama H, Kohno T, Ishii Y, Shimada Y, Shiraishi K, Iwakawa R, Furuta K, Tsuta K, Shibata T, Yamamoto S, et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012;72:100–11.
Yamauchi M, Yamaguchi R, Nakata A, Kohno T, Nagasaki M, Shimamura T, Imoto S, Saito A, Ueno K, Hatanaka Y, et al. Epidermal growth factor receptor tyrosine kinase defines critical prognostic genes of stage I lung adenocarcinoma. PLoS ONE. 2012;7:e43923.
Der SD, Sykes J, Pintilie M, Zhu CQ, Strumpf D, Liu N, Jurisica I, Shepherd FA, Tsao MS. Validation of a histology-independent prognostic gene signature for early-stage, non-small-cell lung cancer including stage IA patients. J Thorac Oncol. 2014;9:59–64.
Schabath MB, Welsh EA, Fulp WJ, Chen L, Teer JK, Thompson ZJ, Engel BE, Xie M, Berglund AE, Creelan BC, et al. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene. 2016;35:3209–16.
Goldman M, Craft B, Swatloski T, Cline M, Morozova O, Diekhans M, Haussler D, Zhu J. The UCSC Cancer Genomics Browser: update 2015. Nucleic Acids Res. 2015;43:D812–817.
Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173(400–416):e411.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, Winsor GL, Hancock RE, Brinkman FS, Lynn DJ. InnateDB: systems biology of innate immunity and beyond–recent updates and continuing curation. Nucleic Acids Res. 2013;41:D1228–1233.
Wang W, Zhao Z, Yang F, Wang H, Wu F, Liang T, Yan X, Li J, Lan Q, Wang J, Zhao J. An immune-related lncRNA signature for patients with anaplastic gliomas. J Neurooncol. 2018;136:263–71.
Zhang W, Zhang J, Yan W, You G, Bao Z, Li S, Kang C, Jiang C, You Y, Zhang Y, et al. Whole-genome microRNA expression profiling identifies a 5-microRNA signature as a prognostic biomarker in Chinese patients with primary glioblastoma multiforme. Cancer. 2013;119:814–24.
He R, Zuo S. A robust 8-gene prognostic signature for early-stage non-small cell lung cancer. Front Oncol. 2019;9:693.
survminer: Drawing Survival Curves using 'ggplot2'. https://CRAN.R-project.org/package=survminer.
A Package for Survival Analysis in S. https://CRAN.R-project.org/package=survival.
Blanche P, Dartigues JF, Jacqmin-Gadda H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat Med. 2013;32:5381–97.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7.
R: A Language and Environment for Statistical Computing. https://www.R-project.org/.
Atchison CM, Arlikar S, Amankwah E, Ayala I, Barrett L, Branchford BR, Streiff M, Takemoto C, Goldenberg NA. Development of a new risk score for hospital-associated venous thromboembolism in noncritically ill children: findings from a large single-institutional case-control study. J Pediatr. 2014;165:793–8.
Kang SJ, Cho YR, Park GM, Ahn JM, Han SB, Lee JY, Kim WJ, Park DW, Lee SW, Kim YH, et al. Predictors for functionally significant in-stent restenosis: an integrated analysis using coronary angiography, IVUS, and myocardial perfusion imaging. JACC Cardiovasc Imaging. 2013;6:1183–90.
pheatmap: Pretty Heatmaps. https://CRAN.R-project.org/package=pheatmap
Brock MV, Hooker CM, Ota-Machida E, Han Y, Guo M, Ames S, Glockner S, Piantadosi S, Gabrielson E, Pridham G, et al. DNA methylation markers and early recurrence in stage I lung cancer. N Engl J Med. 2008;358:1118–28.
Kratz JR, He J, Van Den Eeden SK, Zhu ZH, Gao W, Pham PT, Mulvihill MS, Ziaei F, Zhang H, Su B, et al. A practical molecular assay to predict survival in resected non-squamous, non-small-cell lung cancer: development and international validation studies. Lancet. 2012;379:823–32.
Rakha E, Pajares MJ, Ilie M, Pio R, Echeveste J, Hughes E, Soomro I, Long E, Idoate MA, Wagner S, et al. Stratification of resectable lung adenocarcinoma by molecular and pathological risk estimators. Eur J Cancer. 2015;51:1897–903.
Vargas AJ, Harris CC. Biomarker development in the precision medicine era: lung cancer as a case study. Nat Rev Cancer. 2016;16:525–37.
Xia H, Ma YF, Yu CH, Li YJ, Tang J, Li JB, Zhao YN, Liu Y. Aquaporin 3 knockdown suppresses tumour growth and angiogenesis in experimental non-small cell lung cancer. Exp Physiol. 2014;99:974–84.
Cao Y, Zhu W, Chen W, Wu J, Hou G, Li Y. Prognostic value of BIRC5 in lung adenocarcinoma lacking EGFR, KRAS, and ALK mutations by integrated bioinformatics analysis. Dis Markers. 2019;2019:5451290.
Yi J, Wei X, Li X, Wan L, Dong J, Wang R. A genome-wide comprehensive analysis of alterations in driver genes in non-small-cell lung cancer. Anticancer Drugs. 2018;29:10–8.
Tsai JR, Wang HM, Liu PL, Chen YH, Yang MC, Chou SH, Cheng YJ, Yin WH, Hwang JJ, Chong IW. High expression of heme oxygenase-1 is associated with tumor invasiveness and poor clinical outcome in non-small cell lung cancer patients. Cell Oncol (Dordr). 2012;35:461–71.
Sorrentino C, Di Carlo E. Expression of IL-32 in human lung cancer is related to the histotype and metastatic phenotype. Am J Respir Crit Care Med. 2009;180:769–79.
Brooks GD, McLeod L, Alhayyani S, Miller A, Russell PA, Ferlin W, Rose-John S, Ruwanpura S, Jenkins BJ. IL6 Trans-signaling promotes KRAS-driven lung carcinogenesis. Cancer Res. 2016;76:866–76.
Li J, Zhang J, Xie F, Peng J, Wu X. Macrophage migration inhibitory factor promotes Warburg effect via activation of the NFkappaB/HIF1alpha pathway in lung cancer. Int J Mol Med. 2018;41:1062–8.
Ella E, Harel Y, Abraham M, Wald H, Benny O, Karsch-Bluman A, Vincent D, Laurent D, Amir G, Izhar U, et al. Matrix metalloproteinase 12 promotes tumor propagation in the lung. J Thorac Cardiovasc Surg. 2018;155(2164–2175):e2161.
Zhou J, Kwak KJ, Wu Z, Yang D, Li J, Chang M, Song Y, Zeng H, Lee LJ, Hu J, Bai C. PLAUR confers resistance to gefitinib through EGFR/P-AKT/survivin signaling pathway. Cell Physiol Biochem. 2018;47:1909–24.
Do H, Kim D, Kang J, Son B, Seo D, Youn H, Youn B, Kim W. TFAP2C increases cell proliferation by downregulating GADD45B and PMAIP1 in non-small cell lung cancer cells. Biol Res. 2019;52:35.
Zhou Y, Liao Q, Han Y, Chen J, Liu Z, Ling H, Zhang J, Yang W, Oyang L, Xia L, et al. Rac1 overexpression is correlated with epithelial mesenchymal transition and predicts poor prognosis in non-small cell lung cancer. J Cancer. 2016;7:2100–9.
Zeng Z, Yang Y, Qing C, Hu Z, Huang Y, Zhou C, Li D, Jiang Y. Distinct expression and prognostic value of members of SMAD family in non-small cell lung cancer. Medicine (Baltimore). 2020;99:e19451.
Gachechiladze M, Tichy T, Kolek V, Grygarkova I, Klein J, Mgebrishvili G, Kharaishvili G, Janikova M, Smickova P, Cierna L, et al. Sphingosine kinase-1 predicts overall survival outcomes in non-small cell lung cancer patients treated with carboplatin and navelbine. Oncol Lett. 2019;18:1259–66.
Zhang C, Lu J, Zhang QW, Zhao W, Guo JH, Liu SL, Wu YL, Jiang B, Gao FH. USP7 promotes cell proliferation through the stabilization of Ki-67 protein in non-small cell lung cancer cells. Int J Biochem Cell Biol. 2016;79:209–21.
Ajona D, Zandueta C, Corrales L, Moreno H, Pajares MJ, Ortiz-Espinosa S, Martinez-Terroba E, Perurena N, de Miguel FJ, Jantus-Lewintre E, et al. Blockade of the complement C5a/C5aR1 Axis impairs lung cancer bone metastasis by CXCL16-mediated effects. Am J Respir Crit Care Med. 2018;197:1164–76.
Li R, Peng C, Zhang X, Wu Y, Pan S, Xiao Y. Roles of Arf6 in cancer cell invasion, metastasis and proliferation. Life Sci. 2017;182:80–4.
Hashimoto S, Mikami S, Sugino H, Yoshikawa A, Hashimoto A, Onodera Y, Furukawa S, Handa H, Oikawa T, Okada Y, et al. Lysophosphatidic acid activates Arf6 to promote the mesenchymal malignancy of renal cancer. Nat Commun. 2016;7:10656.
Onodera Y, Hashimoto S, Hashimoto A, Morishige M, Mazaki Y, Yamada A, Ogawa E, Adachi M, Sakurai T, Manabe T, et al. Expression of AMAP1, an ArfGAP, provides novel targets to inhibit breast cancer invasive activities. EMBO J. 2005;24:963–73.
Sabe H. Requirement for Arf6 in cell adhesion, migration, and cancer cell invasion. J Biochem. 2003;134:485–9.
Hashimoto S, Furukawa S, Hashimoto A, Tsutaho A, Fukao A, Sakamura Y, Parajuli G, Onodera Y, Otsuka Y, Handa H, et al. ARF6 and AMAP1 are major targets of KRAS and TP53 mutations to promote invasion, PD-L1 dynamics, and immune evasion of pancreatic cancer. Proc Natl Acad Sci USA. 2019;116:17450–9.
Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57.
Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–46.
Suzuki K, Kadota K, Sima CS, Nitadori J, Rusch VW, Travis WD, Sadelain M, Adusumilli PS. Clinical impact of immune microenvironment in stage I lung adenocarcinoma: tumor interleukin-12 receptor beta2 (IL-12Rbeta2), IL-7R, and stromal FoxP3/CD3 ratio are independent predictors of recurrence. J Clin Oncol. 2013;31:490–8.
Donnem T, Hald SM, Paulsen EE, Richardsen E, Al-Saad S, Kilvaer TK, Brustugun OT, Helland A, Lund-Iversen M, Poehl M, et al. Stromal CD8+ T-cell density-A promising supplement to TNM staging in non-small cell lung cancer. Clin Cancer Res. 2015;21:2635–43.
Acknowledgements
We thank Dr. Jianming Zeng and his biotrainee platform and Dr. Guo for their contributions to the bioinformatics knowledge sharing.
Funding
This work was supported by the Natural Science Foundation of Beijing, China (Grant No.7182132).
Author information
Authors and Affiliations
Contributions
PC W and YZ conceived and designed the study. PC W and YZ analyzed the data and wrote the manuscript. YY W and YD W prepared the figures and tables. PC W and NX L revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
The general information of the enrolled datasets.
Additional file 2: Table S2.
The baseline characteristics of patients included in this study.
Additional file 3: Table S3.
The list of the immune-related genes downloaded from the InnateDB database.
Additional file 4: Table S4.
The detailed information of the system, software and packages using in this study.
Additional file 5: Table S5.
The general information of the overlapped prognostic genes and corresponding coefficients.
Additional file 6: Table S6.
The specific risk score formula using in this study.
Additional file 7: Table S7.
The cutoff values for each data set.
Additional file 8: Figure S1.
The distribution of the risk scores, survival status and gene expression levels in the enrolled data set.
Additional file 9: Table S8.
The data for Kaplan-Meier survival analysis and ROC analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Wu, P., Zheng, Y., Wang, Y. et al. Development and validation of a robust immune-related prognostic signature in early-stage lung adenocarcinoma. J Transl Med 18, 380 (2020). https://doi.org/10.1186/s12967-020-02545-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-020-02545-z