
Abstract
Background
Complement Regulatory Proteins (CRPs), especially CD55 primarily negate complement factor 3-mediated injuries and maintain tissue homeostasis during complement cascade activation. Complement activation and regulation during alloimmune inflammation contribute to allograft injury and therefore we proposed to investigate a crucial pathological link between vascular expression of CD55, active-C3, T cell immunity and associated microvascular tissue injuries during allograft rejection.
Methods
Balb/c→C57BL/6 allografts were examined for microvascular deposition of CD55, C3d, T cells, and associated tissue microvascular impairments during rejection in mouse orthotopic tracheal transplantation.
Results
Our findings demonstrated that hypoxia-induced early activation of HIF-1α favors a cell-mediated inflammation (CD4+, CD8+, and associated proinflammatory cytokines, IL-2 and TNF-α), which proportionally triggers the downregulation of CRP-CD55, and thereby augments the uncontrolled release of active-C3, and Caspase-3 deposition on CD31+ graft vascular endothelial cells. These molecular changes are pathologically associated with microvascular deterioration (low tissue O2 and Blood flow) and subsequent airway epithelial injuries of rejecting allografts as compared to non-rejecting syngrafts.
Conclusion
Together, these findings establish a pathological correlation between complement dysregulation, T cell immunity, and microvascular associated injuries during alloimmune inflammation in transplantation.
Similar content being viewed by others


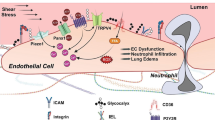
Background
Microvascular loss has been associated with the progression of tissue remodeling and organ rejection during transplantation. Various preclinical and clinical studies have demonstrated a marked loss of microvasculature in nonfibrotic airways a finding suggesting that a loss of microvasculature precedes and may be related to the progression of chronic rejection in transplanted lungs [1,2,3,4,5]. Microvascular loss and associated tissue injuries due to alloimmune inflammation involves an organized communication of myriad of cellular (T cells, B cells, Macrophages, and Dendritic cells) and molecular components (Complement factors and Immunoglobulins) which play a crucial role in the development of tissue remodeling, and progression of small airways closure and subsequent decline in pulmonary functions [1, 2, 6,7,8]. Microvascular loss during complement-mediated inflammation, and subsequent microangiopathy with its attendant ischemia, can lead to tissue infarction and terminal airway fibrosis [1, 2, 5, 9], and therefore, maintaining healthy microvasculature with various immunotherapies in rejecting allografts may be crucial for preventing terminal airway fibrosis [3, 5, 10].
Role of complement and associated regulatory mechanism has been studied in ischemia/reperfusion injury and other associated pathological alterations during transplantation but a direct pathological impact of C3 activation and regulatory proteins is not well known during alloimmune inflammation post-transplantation. Activation of complement cascade release C3, C5 anaphylatoxins, and other toxic peptides, which play a decisive role in pathophysiological injuries during alloimmune inflammation [11,12,13,14,15]. To shield against the complement-mediated injury, host tissues express several membrane-anchored CRPs, which include decay-accelerating factor (DAF; CD55), membrane cofactor protein (MCP), complement receptor 1-related gene/protein y (Crry), complement receptor 1 (CR1) and CD59 [16, 17]. All these CRPs are unique in targeting and operate only at specific steps of complement activation, which makes them highly sophisticated regulatory switch to protect cellular machinery during the extreme inflammatory phase [17,18,19,20,21,22,23].
CD55 inhibits the formation of the C3-convertase, the major amplification step in the complement activation cascade and plays a critical role in negating complement-mediated injury in the pathophysiology of various inflammatory diseases including allograft rejection [24,25,26,27,28,29,30,31,32,33]. Here, we proposed to investigate a possible relationship between the key activated complement factor C3, and CD55, which is normally expressed on vascular endothelium and is the focus of the ongoing research plan. Further, we aim to determine a pathological correlation between graft expression of CD55 and C3d during microvascular rejection in an experimental mouse model of orthotopic tracheal transplant [1, 7, 34].
Methods
Mice
All MHC-mismatched mice selected for this investigation were originally acquired from the Jackson Laboratory (JAX, USA), and preserved as an original breeder colony in our King Faisal Specialist Hospital and Research Centre animal facility (KFSH&RC) at Riyadh, Saudi Arabia. All animal procedures used in this study were approved by KFSH& RC Animal Care and Use Committee (ACUC).
BALB/c
BALB/c (H-2d) mice were selected as an allogeneic tracheal graft donor for the C57BL/6 mice recipients in the orthotopic tracheal transplants.
C57bl/6
C57BL/6 (B6.H-2b) mice were selected as a tracheal graft recipient in all allograft conditions while they were selected as a tracheal graft donor in syngraft conditions as described in the experimental plan (Table 1).
Surgical procedure
The surgical procedure of Orthotopic Tracheal Transplants (OTT) was performed under sterile conditions and different groups were surgically transplanted. In brief, recipient mice were anesthetized (Ketamine 100 mg/kg and Xylazine 20 mg/kg) and trachea was bisected with small incision, and donor tracheal segment of 4–6 rings was surgically sutured into the recipient with 10-0 nylon suture (AROSurgical, USA) at each anterior and posterior end of the bisected trachea. Next, all the surrounding tissue were placed and the overlying skin was closed with 5-0 silk suture [34, 35]. All transplanted mice were given Carprofen (dose 5 mg/kg × SC) and Zolecin (dose 100 mg/kg × SC) and were monitored for any respiratory distress-stridor in the first 24 h after surgery. To examine the rejection and associated microvascular kinetics, allografts were selected at specific days post tracheal transplantation, which starts from d6 (point of early microvascular flow), d8 (point of maximum microvascular flow), d10 (point of microvascular loss/acute rejection) to better see a pathological correlation between expression of active complement factors and CRP-CD55 with associated tissue and microvascular injuries in rejecting allografts [9, 36, 37].
Graft functional microvasculature
To examine the functional microvascular blood flow between donor and recipient grafts, transplanted mice were anesthetized and quickly injected intravenously with 50 μl (1 mg/ml FITC-conjugated Lycopersicon esculentum) of tomato lectin [7] and waited for 5 min before flushing the whole vasculature with 1% PFA (paraformaldehyde) via the aorta. After PFA washing, the graft was harvested and fixed in 1% PFA at 4 °C for 10 min. Next, the graft was mounted and examined by Immunofluorescence microscopy (EVOS FL auto cell imaging system, Life Technologies, USA) to detect the presence of FITC-tagged functional graft microvasculature [9, 37].
Graft oxygenation and blood flow
The oxygen (tpO2 mmHg) and blood perfusion (BPUs) during rejection were measured by combined oxygen and blood flow sensors (model NX-BF/OF/E, Oxford Optronix, UK) as described earlier with some modifications [5, 9, 34, 37]. Briefly, the transplanted mouse was anesthetized and the graft was exposed for oxygen and blood flow measurement. Next, a 23G needle was used to make a hole in the graft, and a combined sensor was gradually inserted through a micromanipulator until it reaches the epithelium of the graft. Since the graft is orthotopic so it always remains in the vicinity of inhaled oxygen, which may interfere with tissue oxygen. To avoid and minimize this inhaled oxygen, the sensor was lowered until the tpO2 levels decrease to 5 mmHg or less (indicating a zeroing effect induced by tissue compression), and subsequently raised in small increments through micromanipulator until the tpO2 and BPU reading plateaus and a consistent reading was obtained [34, 35]. Of note, detachment of the sensor from the tissue epithelium spiked the oxygen content (> 45 mmHg) therefore we routinely optimized this measurement while the sensor remains in firm contact with epithelium, which we regulate through micromanipulator.
Immunofluorescence staining
To demonstrate cellular inflammation, and complement dysregulation, we examined harvested allograft at d6 and d10 post-transplantation and immunostained for CD4+, CD8+ T cells, and vascular endothelial deposition of CD55 and active C3 during post-transplantation. In brief, harvested and Tissue-Tek O.C.T. (Sakura Finetek, Japan) frozen grafts were sliced (5 μm) through a cryostat (HM550; Microm) and the sections were mounted on super frost/plus slides (Fisher Scientific) for immunofluorescence staining as described [3, 5, 9, 37]. After methanol/acetone (1:1) fixation for 10 min at − 20 °C, the slides were washed and incubated with 10% donkey serum for 30 min. Next, slides were overnight incubated with either Rat anti-mouse CD4 (BD biosciences, USA) Rat anti-mouse CD8 (BD biosciences, USA), Rabbit anti-mouse CD55 (Abcam, USA), Goat anti-mouse C3 (R&D systems, USA), Rabbit anti-mouse Caspase-3 (R&D Systems, USA) and Rat anti-mouse CD31 (R&D Systems, USA) specific primary antibodies, and slides were incubated overnight and later rinsed (3×) gently to remove any non-specific binding. Next, slides were incubated with Cy3 donkey anti-rat (Jackson ImmunoResearch Laboratories, USA), Alexa 488 goat anti-rabbit (Jackson ImmunoResearch Laboratories, USA) or with Alexa 488 Donkey anti-goat (Jackson ImmunoResearch Laboratories, USA) secondary antibodies for 1 h. at 37 °C. After incubation, slides were gently rinsed (3×) and mounted in Vectashield mounting medium (Vector Laboratories, USA) for image acquisition. Image acquisition and morphometric analysis of selected markers were performed on randomly selected high-powered fields on EVOS FL auto cell imaging system (Life Technologies, USA), and the percent co-localization of two markers was quantified as mean integrated fluorescence intensity using ImageJ program [7, 9, 37].
Flow cytometry
To isolate CD55 expressing vascular endothelial cells, collagenase digestion of harvested graft was performed according to the standard protocol with some modifications [38]. Briefly, each freshly harvested graft was minced in cold HEPES buffer, and incubated with 2.5 mg/ml of collagenase D (11088858001 Roche, USA) for 1 h. at room temperature in 50RPM shaking bath. After incubation, the cell suspension was filtered with 70um cell strainers (Falcon, USA), and centrifuged at 400g for 30 min. Next, the isolated cell population was stained for surface expression of CD55 on CD31+ endothelial cells, and Flow Cytometric analysis was performed at the flow rate of 14 μl/min and a minimum of 500,000 events. All data were later analyzed through BD Accuri C6 integrated software version C6 [5]. Further, to examine the circulating T effector cells (CD4+ and CD8+) expression during rejection, blood samples were collected (BD-vacutainers) and lymphocyte buffy coat was separated through Hisptopaque gradient procedure as described [3, 5, 39], and mouse T effector specific markers were stained with APC-conjugated anti-mouse CD4+ (Clone RM4-5 RUO, BD Pharmingen), and Alexa488-conjugated CD8+ (Clone 53-6.7, BD Pharmingen) respectively as recommended by BD Pharmingen assay, which specifically flow sort CD4+ and CD8+ in a given lymphocytes population. Data were recorded at the flow rate of 14ul/min and a minimum of 500,000 events were collected, and further analyzed through BD Accuri C6 integrated software [3, 5].
Histopathology
Pathological changes in the airway epithelium of allografts were evaluated by H&E staining as described [3, 5, 40]. In brief, harvested and Tissue-Tek O.C.T. medium (Sakura Finetek, Japan) processed graft sections on super frost/plus slides (Fisher Scientific) were stained by H&E to detect any pathological and structural perturbations in airway epithelium and mononuclear cell infiltration [9, 41].
Quantitative PCR
RT-PCR analysis of mRNA expression of HIF-1α, CD55, and key inflammatory cytokines was performed with some modifications [3, 5, 9]. Briefly, total RNA from tracheal graft was extracted using RNeasy mini kit 50 (Qiagen Sciences, Maryland, USA.) and quantified using a NanoDrop 1000 spectrophotometer (NanoDrop Technologies, USA). cDNA from each isolated RNA was synthesized with a Superscript™ II cDNA reverse transcription kit (ThermoFisher Scientific) and real time-PCR was performed using gene-specific primers on an AB 7500 Fast Real-Time PCR system in triplicates using Power SYBR Green (ThermoFisher Scientific). Data were analyzed with integrated software, and expression levels were analyzed by the 2−∆∆Ct method after normalization to the housekeeping genes glutaraldehyde dehydrogenase (GAPDH). We selected the Hypoxia-inducible gene (HIF1-α), CD55, and T cell-specific (IL-2 and TNF-α) gene transcripts for the detection of hypoxic, regulatory and inflammatory phases of graft. The sequence of individual primers used in the present study is shown in Table 2.
Statistical analysis
Statistical comparison between groups was performed using two-way ANOVA with post hoc Bonferroni correction for multiple comparisons while single comparisons were analyzed by 1-way ANOVA through GraphPad™ Prism software. A p-value < 0.05 was considered significant.
Results
Loss of graft functional microvasculature during rejection
While the deleterious effects of C3 deposition on vascular reestablishment, tissue oxygenation, and blood perfusion had already been reported earlier [9], here we further delineated a fine correlation between CD55/C3d balance, airway microvasculature and epithelial injuries during allograft rejection. As reported in previous transplant settings, activation of the complement cascade and associated complement regulatory protein dysregulation has been a key to affect microvascular blood flow, tissue oxygenation and airway epithelial repair during airway allograft rejection [3, 42]. We hypothesized that vascular inflammation is directly associated with donor-recipient microvasculature, and microvascular flow and tissue oxygenation. In this study, allograft was monitored for tissue oxygenation, microvascular blood perfusion, and the occurrence of donor-recipient microvasculature in the course of airway rejection.
First, to demonstrate functional microvasculature between the donor and recipient tracheal grafts, we transplanted C57BL/6 recipients with tracheas from MHC-incompatible BALB/c donors (allografts), and harvested trachea at selected time point post-transplantation. To check the microvasculature, rejecting allografts were examined by FITC-lectin binding assay, which specifically maps the existence of functional microvasculature between donor and recipient grafts [9, 31, 37]. We found that allograft remains perfused from d6 to d8 but lost microvascular perfusion at d10 and remained unperfused (Fig. 1a–c). Next, we examined the levels of tissue oxygenation and microvascular blood flow (measured in blood perfusion units, BPUs) in rejecting allografts and in syngraft at d6, d8, and d10 post-transplantation. Our results demonstrate that BALB/c→C57BL/6 allografts remain oxygenated from d6–d8 but pass through a period of hypoxia and ischemia on d10 post-transplantation while syngraft remains oxygenated during this phase (Fig. 1a–c). Next, we examined harvested grafts for HIF-1α mRNA expression during rejection in rejecting allografts. We found a significant early increase in HIF-1α mRNA expression in allograft compared to syngraft controls, which further established a pathological correlation that microvascular injury coincides with the onset of the hypoxic state which eventually triggers higher HIF-1α expression in allograft compared to their control syngraft (Fig. 1d).

Allograft rejection is associated with donor-recipient microvasculature loss. a Lectin binding assay of whole-mount tracheal grafts on d6, d8, and d10. Original magnification, ×20. b, c Tissue pO2 (Mean ± SE, mmHg) and Blood perfusion (Mean ± SE, units) were plotted over different time points. d Quantitative RT-PCR analysis shows fold change in HIF1-α mRNA at d6 post-transplantation. Data are presented as means with SE of 6–12 transplants/time point/experiment. *p < 0.05
Microvascular loss is associated with cellular inflammation
Here, we tested the alloimmune response of peripheral and graft infiltration of CD4+ and CD8+ T cells during the phase of acute rejection, which is defined by the loss of microvasculature and subsequent hypoxic state. To investigate the inflammatory state during microvascular loss, we surgically transplanted MHC-incompatible C57BL/6 (B6, H-2b) with tracheas of BALB/c (H-2d) donors. Next, blood lymphocytes were isolated from BALB/c (H-2d)→C57BL/6 (H-2b) control allografts and C57BL/6 (H-2b)→C57BL/6 (H-2b) syngraft for peripheral CD4+, CD8+ T effector cells at d6 and d10 post-transplantation. We found that allografts exhibited a significant increase in CD4+ and CD8+ T cells both in peripheral blood and graft, compared to syngraft control at d6 and d10 post-transplantation (Fig. 2a, b). These findings indicate that functional microvascular loss during rejection is associated with the increase in both peripheral and graft infiltration of CD4+ and CD8+ T cells. Collectively, this creates a favorable inflammatory environment to initiate and augment microvascular associated injuries, which play a crucial role in the development of airway tissue remodeling during transplantation. To further confirm the inflammatory state, we performed RT-PCR analysis of two key inflammatory cytokines (IL-2 and TNF-α) in harvested grafts at d10 post-transplantation (Fig. 1c). Our mRNA expression analysis demonstrated that T cell-specific cellular infiltration is associated with an influx of proinflammatory cytokines, which will play a decisive role in tissue microvascular injury during immune rejection.

Microvascular loss is associated with an increase in graft T cells infiltration: a, b Immunofluorescent staining for CD4+, and CD8+ at 10 post-transplantation. c Quantitative RT-PCR analysis shows fold change in IL-2 and TNF-α mRNA at d10 post-transplantation. d Flow cytometry analysis of peripheral lymphocytes (CD4+, and CD8+) at d10 post-transplantation. Data are presented as means with SE of 6–12 transplants/time point/experiment. *p < 0.05
Microvascular loss is associated with complement dysregulation
To avoid complement-mediated injury, host tissues express a number of membrane-anchored CRPs including decay-accelerating factor (DAF; CD55). To investigate the complement dysregulation and potential complement-mediated graft microvascular injuries during transplantation, tracheal allografts (BALB/c→B6) were examined through mRNA expression, flow cytometry and immunofluorescence imaging at d6, and d10 post-transplantation.
To evaluate the downregulation of CD55 on graft microvascular stratum, we first harvested allografts and syngrafts at d6 and d10 post-transplantation and tested the mRNA expression of CD55. The pattern of mRNA expression showed a significant low CD55 mRNA expression in both d6 and d10 allografts as compared to the corresponding syngraft. To further confirm the CD55 expression during rejection, we harvested both allograft and syngraft and processed them for collagenase digestion assay. The liberated vascular endothelial cells after collagenase treatment were filtered and immunostained for surface expression of CD55 and CD31 proteins. The acquired immunostained cells through flow cytometry exhibited a lower expression of CD55 on CD31+ vascular endothelial cells at d10 post-transplantation in allografts as compared to syngraft controls. In addition, both allograft and syngraft were further examined through immunofluorescence staining at d10 (point of microvascular loss) post-transplantation. Image analysis of CD55 and CD31 coexpression further revealed a downward trend in CD55 expression on CD31+ vascular endothelial cells at d6 and d10 post-transplantation. These findings suggest that vascular endothelial expression of CD55 down-regulated during allograft rejection at d10 post-transplantation which coincides with the loss of microvasculature (Fig. 3a–c).

Microvascular loss is associated with complement dysregulation on vascular endothelial cells. a Quantitative RT-PCR analysis shows fold change in CD55 mRNA at d6 and d10 post-transplantation. b Flow cytometry analysis of vascular endothelial cells isolated from collagenase digested allograft and syngraft at d10 post-transplantation. c Immunofluorescent staining for CD55, and CD31 at d6, and 10 post-transplantation in rejecting allografts. d Immunofluorescent staining for C3d, and CD31 at d6, and 10 post-transplantation in rejecting allografts. Data are presented as means with SE of 6–12 transplants/time point/experiment. *p < 0.05. Original magnification, ×40
To investigate the correlation of CD55 downregulation on associated complement activation, we next examined the deposition of activated complement factor C3d on vascular endothelial cells at d6 and d10 post-transplantation. Our immunofluorescence data demonstrated an upward trend in C3d deposition on CD31+ vascular endothelial cells at d6-d10 post-transplantation. These findings suggested that vascular endothelial deposition of active C3 is a major molecular signature of tissue microvascular injury during rejection. These findings support the notion that d10, which is the point of microvascular loss coincides with the low deposition of CD55 and high deposition of C3d on vascular endothelial cells as seen in rejecting allograft compare to syngrafts (Fig. 3a–d). Of note, the syngrafts do not show any significant increase in both CD55 and C3d expression on CD31+ vascular endothelial cells.
Microvascular loss is associated with endothelial caspase-3 activation
Allografts endothelial cells are a primary target of vascular injury during cytotoxic T lymphocytes (CTL) mediated alloreactivity, which greatly impacts microvascular health and progressive vascular loss. To determine the vascular expression of CTL mediated caspase-3 activation, harvested allograft, and syngraft sections were immunostained for Caspase-3 deposition on vascular endothelial cells at d10 post-transplantation, which coincides with the CD8+ T cells infiltration in the graft. Immunofluorescence staining demonstrated a significant increase in Caspase-3 on CD31+ vascular endothelial cells during inflammation. These findings suggested that vascular endothelial deposition of active Caspase-3 is a key apoptotic factor generated due to CTL mediated alloreactivity during microvascular injury (Fig. 4).

Microvascular loss is associated with Caspase-3 activation on vascular endothelial cells. Immunofluorescent staining for Caspase-3 and CD31 at d10 post-transplantation. Data are presented as means of 12 transplants/time point/experiment. *p < 0.05. Original magnification, ×40
Microvascular loss is associated with airway epithelial injury
Airway epithelial injuries are the leading pathological event, and key irreversible pathological consequences that proceed when rejecting allografts undergo a state of severe hypoxia/ischemia, and ultimately to airway remodeling during the terminal phase of rejection [37]. To examine airway epithelial injuries, slides were stained by H&E to detect airway epithelial structures in allografts and syngrafts at d6 and d10 post-transplantation as described previously [40] and stained graft slices were imaged using a Leica light microscope to localize epithelial and subepithelial zones. Microscopic examinations of H&E showed an inflamed airway epithelium with massive infiltration of mononuclear cells in subepithelial tissues at d6 post-transplantation, while d10 allograft showed a partial or complete loss of airway epithelium but subepithelial tissues remain populated with massive mononuclear cell infiltration [9, 41]. However, corresponding syngrafts showed no sign of airway epithelial injury or infiltration of subepithelial mononuclear cells during transplantation (Fig. 5).

Microvascular loss is associated with an epithelial injury. H&E staining of BALB/c→C57BL/6 allografts at d6, and d10 post-transplantation. SE designate graft sub-epithelial area. Data are presented as means of 12 transplants/time point/experiment. *p < 0.05. Original magnification, ×40
Discussion
CRPs are the crucial regulatory switch available to check the uncontrolled active complement deposition and negate their toxic effects on graft vasculature during alloimmune reactions [43, 44]. CRPs, including CD55, complement receptor 1-related gene/protein y (Crry) and CD59 usually modulate deleterious effects of active complement mediators during allograft rejection [21, 28]. Complement accumulation on the vascular endothelium precedes the development of airway fibrosis and subsequent destruction of blood vessels and microvascular blood flow to the rejecting allograft. In addition, previous findings have strengthened the fact that complement deficiency or blockade leads to markedly improved oxygenation in rejecting allografts [5, 9, 37]. To date, work focusing on the impact of hypoxia on CRP expression has been limited to in vitro studies [45, 46]. While these studies have been important in showing how CRPs can be upregulated in cultured cells in hypoxic conditions, they cannot replicate the complex in vivo milieu of a rejecting transplant [47]. Inverse correlation of tissue expression of active complement components and CRPs have been crucial to the progress of transplant injury and associated tissue remodeling during rejection [48,49,50].
The key hypothesis to be tested in this manuscript is that during alloimmune inflammation, graft microvasculature and the airway epithelium is rendered susceptible to complement-mediated injury through decreased expression of major complement regulatory protein CD55 during a mouse model of OTT. The OTT model is a well-established model to investigate the complement-mediated graft ischemia and airway tissue remodeling during rejection [5, 9, 37]. This study was, therefore, the first to examine the vascular expression of CD55, C3d kinetics, and T cell immunity, and how it correlated to the development of microvascular loss and airway tissue remodeling.
Herein, we demonstrated that early hypoxic state favors a cell-mediated inflammation, which proportionally triggers the downregulation of CD55, and thereby augments the uncontrolled release of active-C3, and Caspase-3 deposition on graft vascular endothelial cells. As reported in previous preclinical studies, alloimmune inflammation is characterized by a massive T cell infiltration (CD4+ and CD8+), associated proinflammatory cytokines, vascular deposition of CTL mediated Caspase-3 activation, which mutually take part in tissue and microvascular damage [9, 15, 37]. Our preliminary results also demonstrated a significant increase in early activation of HIF-1α mRNA (d6), followed by a massive increase in T cell infiltration and major proinflammatory cytokines (IL-2 and TNF-α) during microvascular deterioration. These findings supported a notion that inflammation-mediated microvascular injury determines hypoxia/ischemia state and consequently led to the early activation of HIF-1α which plays a vital role in the metabolic switches that favor cellular adaptation to hypoxic state [51,52,53]. To further dissect the impact of HIF-1α on complement regulatory protein CD55 on vascular endothelial cells, we examined rejecting allografts by mRNA expression, immunofluorescence, and flow cytometry experiments at d10 (point of acute rejection) to confirm if the vascular expression of CD55 and activated complement C3 corresponds to microvascular associated injuries during alloimmune inflammation. We observed that complement-mediated alloimmune inflammation and associated regulatory machinery (e.g. CD55) showed an inverse pathological relationship during airway inflammation. Furthermore, we found that vascular endothelial expression of C3d increases, while vascular endothelial expression of CD55 shows a corresponding decrease during allograft rejection. Next, we further evaluated the microvascular state of graft during the downregulation of CD55/upregulation of C3d. Our results indicate that C3d mediated alloimmune inflammation and corresponding downregulation of CD55 showed a strong pathological correlation with alloimmune inflammation (CD4+ and CD8+), and associated tissue microvascular injuries, graft hypoxia, ischemia, and airway epithelial loss during airway rejection. Altogether, this unfavorable environment seems to contribute to a proinflammatory and more vascular disruption during an alloimmune inflammation and may help to establish a state of severe hypoxia and ischemia. This hypoxic state triggers an inflammatory response, which follows the recruitment of immune cells and associated proinflammatory cytokines. The role of hypoxia has been extensively reported in various immunological conditions including transplantation, and the occurrence of hypoxia has been narrated as a key phase that affects inflammation during the molecular, and cellular checkpoints [54, 55]. It has been well reported that HIF-1α, as the master regulator of oxygen homeostasis, is an important modulator of the tissue repair phase through its signaling in cell migration, cell survival, cell division, growth factor release, and matrix synthesis during the repair [56, 57]. In addition, hypoxia results in tissue ischemia and the expression of a pro-inflammatory state through the downregulation of complement regulatory protein, CD55 [50]. The crucial role of pathological remodeling during persistent inflammation in small airways has been reported in both preclinical and clinical transplantation data, which obstruct the regeneration of airway epithelium and promote fibro-proliferation due to aberrant tissue repair [58,59,60,61,62]. These epithelial injuries have been recognized as a key intermediate step that is ultimately leading to obliterative airway disease [58,59,60,61]. As reported earlier, C3 in the first major active fragment, which has been reported to initiate microvascular associated injuries through the graft vascular-endothelial deposition [5, 9, 13, 15, 37, 63]. In addition, endothelial deposition of C3 leads to the onset of vascular leakiness, microvascular congestion, thrombus formation, pulmonary edema and neutrophilic invasion of the microvasculature through the interaction of various inflammatory cells [15, 64]. Complement-mediated vascular injury contributes to the pathophysiology of multiple diseases including ischemia–reperfusion injury, myocardial infarction, and various transplantation conditions [65,66,67]. Complement has been recognized as a key regulator of adaptive immunity and various activated fragments have been associated with allograft injury through multiple mechanisms, which include post-transplant IR injury, microvascular injury, alloantibody formation, differentiation and function of alloreactive T cells, and contributes to chronic allograft dysfunctions [27, 28, 68,69,70,71,72,73,74,75,76,77,78]. CD55 is a central complement activation regulator that checks the complement activation through the assembly of C3 convertase (C3bBb), and play a key role in various protective and regulatory impacts of CD55 in various transplant models [23, 27, 79,80,81].
In summary, our results conclude that activation of the complement pathway and corresponding downregulation of CD55 during an alloimmune inflammation are one of the main players in microvascular associated allograft injury. The delineation of this pathological correlation between CRP, activated C3 and tissue remodeling favors the possibility that targeted blocking of activated C3 or the use of recombinant CD55 could be a good therapeutic combination to support microvascular reestablishment during allograft rejection.
Conclusion
Taken together, these findings highlight the key modulatory effects of complement on microvascular associated injuries, and demonstrate a proof-of-concept that targeting C3 blockade could facilitate CD55-mediated vascular protection to allografts. This comparative analysis collectively emphasized that complement activation during alloimmune inflammation is harmful to transplanted tissue, in part, because they contribute to the progression of hypoxia/ischemia by disconnecting microvascular flow between donor-recipient grafts and thus favor hypoxic state. Further, these findings demonstrated a detailed insight of alloimmune response and a fine pathological correlation between complement dysregulation and associated microvascular injuries during rejection. Altogether, this could provide a pathological tool to understand the ongoing pathological state of rejecting graft, and also to better design an anti-complement therapy aimed at preventing microvascular and epithelial injury during alloimmune inflammation.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on request.
Abbreviations
- BPU:
-
Blood perfusion units
- BOS:
-
Bronchiolitis obliterans syndrome
- CRP:
-
Complement regulatory proteins
- D:
-
Day
- DAF (CD55):
-
Decay accelerating factor
- MCP:
-
Membrane cofactor protein
- CTL:
-
Cytotoxic T lymphocyte
- Crry:
-
Complement receptor 1-related gene/protein y
- CR1:
-
Complement receptor 1
- OTT:
-
Orthotopic tracheal transplant model
- PFA:
-
Paraformaldehyde
- Tregs:
-
Regulatory T cells
- tO2 :
-
Tissue oxygen
- O.C.T:
-
Optimal cutting temperature
References
Luckraz H, Goddard M, McNeil K, Atkinson C, Charman SC, Stewart S, Wallwork J. Microvascular changes in small airways predispose to obliterative bronchiolitis after lung transplantation. J Heart Lung Transplant. 2004;23:527–31.
Luckraz H, Goddard M, McNeil K, Atkinson C, Sharples LD, Wallwork J. Is obliterative bronchiolitis in lung transplantation associated with microvascular damage to small airways? Ann Thorac Surg. 2006;82:1212–8.
Khan MA, Alanazi F, Ahmed HA, Al-Mohanna FH, Assiri AM, Broering DC. FOXP3+ regulatory T cell ameliorates microvasculature in the rejection of mouse orthotopic tracheal transplants. Clin Immunol. 2017;174:84–98.
Khan MA, Alanazi F, Ahmed HA, Shamma T, Kelly K, Hammad MA, Alawad AO, Assiri AM, Broering DC. iPSC-derived MSC therapy induces immune tolerance and supports long-term graft survival in mouse orthotopic tracheal transplants. Stem Cell Res Ther. 2019;10:290.
Khan MA, Alanazi F, Ahmed HA, Vater A, Assiri AM, Broering DC. C5a blockade increases regulatory T cell numbers and protects against microvascular loss and epithelial damage in mouse airway allografts. Front Immunol. 2018;9:1010.
Dhillon GS, Zamora MR, Roos JE, Sheahan D, Sista RR, Van der Starre P, Weill D, Nicolls MR. Lung transplant airway hypoxia: a diathesis to fibrosis? Am J Respir Crit Care Med. 2010;182:230–6.
Babu AN, Murakawa T, Thurman JM, Miller EJ, Henson PM, Zamora MR, Voelkel NF, Nicolls MR. Microvascular destruction identifies murine allografts that cannot be rescued from airway fibrosis. J Clin Investig. 2007;117:3774–85.
Jiang X, Sung YK, Tian W, Qian J, Semenza GL, Nicolls MR. Graft microvascular disease in solid organ transplantation. J Mol Med. 2014;92:797–810.
Khan MA, Maasch C, Vater A, Klussmann S, Morser J, Leung LL, Atkinson C, Tomlinson S, Heeger PS, Nicolls MR. Targeting complement component 5a promotes vascular integrity and limits airway remodeling. Proc Natl Acad Sci USA. 2013;110:6061–6.
Heim C, Khan MA, von Silva-Tarouca B, Kuckhahn A, Stamminger T, Ramsperger-Gleixner M, Nicolls MR, Weyand M, Ensminger SM. Preservation of microvascular integrity in murine orthotopic tracheal allografts by clopidogrel. Transplantation. 2019;103:899–908.
Khan MA, Assiri AM, Broering DC. Complement mediators: key regulators of airway tissue remodeling in asthma. J Transl Med. 2015;13:272.
Khan MA, Assiri AM, Broering DC. Complement and macrophage crosstalk during process of angiogenesis in tumor progression. J Biomed Sci. 2015;22:58.
Khan MA, Nicolls MR. Complement-mediated microvascular injury leads to chronic rejection. Adv Exp Med Biol. 2013;734:233–46.
Khan MA, Nicolls MR, Surguladze B, Saadoun I. Complement components as potential therapeutic targets for asthma treatment. Respir Med. 2014;108:543–9.
Khan MA, Shamma T. Complement factor and T-cell interactions during alloimmune inflammation in transplantation. J Leukoc Biol. 2019;105(4):681–94. https://doi.org/10.1002/JLB.5RU0718-288R.
Fischetti F, Tedesco F. Cross-talk between the complement system and endothelial cells in physiologic conditions and in vascular diseases. Autoimmunity. 2006;39:417–28.
Song WC. Complement regulatory proteins and autoimmunity. Autoimmunity. 2006;39:403–10.
Nangaku M. Complement regulatory proteins: are they important in disease? J Am Soc Nephrol. 2003;14:2411–3.
Kawano M. Complement regulatory proteins and autoimmunity. Arch Immunol Ther Exp. 2000;48:367–72.
Kwan WH, van der Touw W, Heeger PS. Complement regulation of T cell immunity. Immunol Res. 2012;54:247–53.
Budding K, van de Graaf EA, Kardol-Hoefnagel T, Broen JC, Kwakkel-van Erp JM, Oudijk EJ, van Kessel DA, Hack CE, Otten HG. A promoter polymorphism in the CD59 complement regulatory protein gene in donor lungs correlates with a higher risk for chronic rejection after lung transplantation. Am J Transplant. 2016;16:987–98.
Piccoli AK, Alegretti AP, Schneider L, Lora PS, Xavier RM. Expression of complement regulatory proteins CD55, CD59, CD35, and CD46 in rheumatoid arthritis. Rev Bras Reumatol. 2011;51:503–10.
Ma KN, Cashman SM, Sweigard JH, Kumar-Singh R. Decay accelerating factor (CD55)-mediated attenuation of complement: therapeutic implications for age-related macular degeneration. Investig Ophthalmol Vis Sci. 2010;51:6776–83.
Heeger PS, Lalli PN, Lin F, Valujskikh A, Liu J, Muqim N, Xu Y, Medof ME. Decay-accelerating factor modulates induction of T cell immunity. J Exp Med. 2005;201:1523–30.
Lalli PN, Strainic MG, Lin F, Medof ME, Heeger PS. Decay accelerating factor can control T cell differentiation into IFN-gamma-producing effector cells via regulating local C5a-induced IL-12 production. J Immunol. 2007;179:5793–802.
Dho SH, Lim JC, Kim LK. Beyond the Role of CD55 as a Complement Component. Immune Netw. 2018;18(1):e11. https://doi.org/10.4110/in.2018.18.e11.
Pavlov V, Raedler H, Yuan S, Leisman S, Kwan WH, Lalli PN, Medof ME, Heeger PS. Donor deficiency of decay-accelerating factor accelerates murine T cell-mediated cardiac allograft rejection. J Immunol. 2008;181:4580–9.
Bongoni AK, Lu B, Salvaris EJ, Roberts V, Fang D, Mcrae JL, Fisicaro N, Dwyer KM, Cowan PJ. Overexpression of human CD55 and CD59 or treatment with human CD55 protects against renal ischemia-reperfusion injury in mice. J Immunol. 2017;198:4837–45.
Nauser CL, Farrar CA, Sacks SH. Complement recognition pathways in renal transplantation. J Am Soc Nephrol. 2017;28:2572–9.
Cravedi P, Heeger PS. Complement as a multifaceted modulator of kidney transplant injury. J Clin Investig. 2014;124:2348–54.
Cravedi P, Leventhal J, Lakhani P, Ward SC, Donovan MJ, Heeger PS. Immune cell-derived C3a and C5a costimulate human T cell alloimmunity. Am J Transplant. 2013;13:2530–9.
Cravedi P, van der Touw W, Heeger PS. Complement regulation of T-cell alloimmunity. Semin Nephrol. 2013;33:565–74.
Liu J, Miwa T, Hilliard B, Chen Y, Lambris JD, Wells AD, Song WC. The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J Exp Med. 2005;201:567–77.
Khan MA, Dhillon G, Jiang X, Lin YC, Nicolls MR. New methods for monitoring dynamic airway tissue oxygenation and perfusion in experimental and clinical transplantation. Am J Physiol Lung Cell Mol Physiol. 2012;303:L861–9.
Hsu JL, Khan MA, Sobel RA, Jiang X, Clemons KV, Nguyen TT, Stevens DA, Martinez M, Nicolls MR. Aspergillus fumigatus invasion increases with progressive airway ischemia. PLoS ONE. 2013;8:e77136.
Jiang X, Khan MA, Tian W, Beilke J, Natarajan R, Kosek J, Yoder MC, Semenza GL, Nicolls MR. Adenovirus-mediated HIF-1alpha gene transfer promotes repair of mouse airway allograft microvasculature and attenuates chronic rejection. J Clin Investig. 2011;121:2336–49.
Khan MA, Jiang X, Dhillon G, Beilke J, Holers VM, Atkinson C, Tomlinson S, Nicolls MR. CD4+ T cells and complement independently mediate graft ischemia in the rejection of mouse orthotopic tracheal transplants. Circ Res. 2011;109:1290–301.
Angyal A, Szekeres Z, Balogh P, Neer Z, Szarka E, Virag V, Medgyesi D, Prechl J, Sarmay G. CD16/32-specific biotinylated 2.4G2 single-chain Fv complexed with avidin–FITC enhances FITC-specific humoral immune response in vivo in a CD16-dependent manner. Int Immunol. 2010;22:71–80.
Bain B, Pshyk K. Enhanced reactivity in mixed leukocyte cultures after separation of mononuclear cells on Ficoll-Hypaque. Transplant Proc. 1972;4:163–4.
Swaisgood CM, French EL, Noga C, Simon RH, Ploplis VA. The development of bleomycin-induced pulmonary fibrosis in mice deficient for components of the fibrinolytic system. Am J Pathol. 2000;157:177–87.
Reinhardt AK, Bottoms SE, Laurent GJ, McAnulty RJ. Quantification of collagen and proteoglycan deposition in a murine model of airway remodelling. Respir Res. 2005;6:30.
Khan MA, Alanazi F, Ahmed HA, Hasan AF, Altuhami A, Assiri AM, Broering DC. The therapeutic potential of Treg cells in preserving microvascular health in a mouse model of orthotopic tracheal transplantation. J Clin Cell Immunol. 2016;7:89.
Michielsen LA, Budding K, Drop D, van de Graaf EA, Kardol-Hoefnagel T, Verhaar MC, van Zuilen AD, Otten HG. Reduced expression of membrane complement regulatory protein CD59 on leukocytes following lung transplantation. Front Immunol. 2008;2017:8.
Xiao K, Fang Z, Gao X, Zhao J, Huang R, Xie M. Membrane complement regulatory protein reduces the damage of transplanting autologous bone marrow mesenchymal stem cells by suppressing the activation of complement. Injury. 2017;48:2089–94.
Kinderlerer AR, Steinberg R, Johns M, Harten SK, Lidington EA, Haskard DO, Maxwell PH, Mason JC. Statin-induced expression of CD59 on vascular endothelium in hypoxia: a potential mechanism for the anti-inflammatory actions of statins in rheumatoid arthritis. Arthritis Res Ther. 2006;8:R130.
Hollborn M, Ackmann C, Kuhrt H, Doktor F, Kohen L, Wiedemann P, Bringmann A. Osmotic and hypoxic induction of the complement factor C9 in cultured human retinal pigment epithelial cells: regulation of VEGF and NLRP3 expression. Mol Vis. 2018;24:518–35.
Yang P, Tyrrell J, Han I, Jaffe GJ. Expression and modulation of RPE cell membrane complement regulatory proteins. Investig Ophthalmol Vis Sci. 2009;50:3473–81.
Nunez K, Thevenot P, Alfadhli A, Cohen A. Complement activation in liver transplantation: role of donor macrosteatosis and implications in delayed graft function. Int J Mol Sci. 2018;19:1750.
Cernoch M, Viklicky O. Complement in kidney transplantation. Front Med. 2017;4:66.
Pandya PH, Fisher AJ, Mickler EA, Temm CJ, Lipking KP, Gracon A, Rothhaar K, Sandusky GE, Murray M, Pollok K, et al. Hypoxia-inducible factor-1alpha regulates CD55 in airway epithelium. Am J Respir Cell Mol Biol. 2016;55:889–98.
Cramer T, Johnson RS. A novel role for the hypoxia inducible transcription factor HIF-1alpha: critical regulation of inflammatory cell function. Cell Cycle. 2003;2:192–3.
Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci USA. 1993;90:4304–8.
Smith SF, Hosgood SA, Nicholson ML. Ischemia–reperfusion injury in renal transplantation: 3 key signaling pathways in tubular epithelial cells. Kidney Int. 2019;95:50–6.
Bartels K, Grenz A, Eltzschig HK. Hypoxia and inflammation are two sides of the same coin. Proc Natl Acad Sci USA. 2013;110:18351–2.
Frohlich S, Boylan J, McLoughlin P. Hypoxia-induced inflammation in the lung: a potential therapeutic target in acute lung injury? Am J Respir Cell Mol Biol. 2013;48:271–9.
Hong WX, Hu MS, Esquivel M, Liang GY, Rennert RC, McArdle A, Paik KJ, Duscher D, Gurtner GC, Lorenz HP, Longaker MT. The role of hypoxia-inducible factor in wound healing. Adv Wound Care. 2014;3:390–9.
Reichner JS, Albina JE. Determination of the role of hypoxia-inducible factor 1 in wound healing. Methods Enzymol. 2004;381:527–38.
Okazaki M, Gelman AE, Tietjens JR, Ibricevic A, Kornfeld CG, Huang HJ, Richardson SB, Lai J, Garbow JR, Patterson GA, et al. Maintenance of airway epithelium in acutely rejected orthotopic vascularized mouse lung transplants. Am J Respir Cell Mol Biol. 2007;37:625–30.
Smith CR, Jaramillo A, Duffy BF, Mohanakumar T. Airway epithelial cell damage mediated by antigen-specific T cells: implications in lung allograft rejection. Hum Immunol. 2000;61:985–92.
Lentsch AB, Ward PA. Regulation of experimental lung inflammation. Respir Physiol. 2001;128:17–22.
Fernandez FG, Jaramillo A, Chen C, Liu DZ, Tung T, Patterson GA, Mohanakumar T. Airway epithelium is the primary target of allograft rejection in murine obliterative airway disease. Am J Transplant. 2004;4:319–25.
Belperio JA, Weigt SS, Fishbein MC, Lynch JP 3rd. Chronic lung allograft rejection: mechanisms and therapy. Proc Am Thorac Soc. 2009;6:108–21.
Seya T, Hara T, Matsumoto M, Sugita Y, Akedo H. Complement-mediated tumor cell damage induced by antibodies against membrane cofactor protein (MCP, CD46). J Exp Med. 1990;172:1673–80.
Khan MA, Hsu JL, Assiri AM, Broering DC. Targeted complement inhibition and microvasculature in transplants: a therapeutic perspective. Clin Exp Immunol. 2016;183(2):175–86. https://doi.org/10.1111/cei.12713.
Atkinson C, He S, Morris K, Qiao F, Casey S, Goddard M, Tomlinson S. Targeted complement inhibitors protect against posttransplant cardiac ischemia and reperfusion injury and reveal an important role for the alternative pathway of complement activation. J Immunol. 2010;185:7007–13.
Austen WG Jr, Zhang M, Chan R, Friend D, Hechtman HB, Carroll MC, Moore FD Jr. Murine hindlimb reperfusion injury can be initiated by a self-reactive monoclonal IgM. Surgery. 2004;136:401–6.
Banda NK, Thurman JM, Kraus D, Wood A, Carroll MC, Arend WP, Holers VM. Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen-induced arthritis. J Immunol. 2006;177:1904–12.
Atkinson C, Song H, Lu B, Qiao F, Burns TA, Holers VM, Tsokos GC, Tomlinson S. Targeted complement inhibition by C3d recognition ameliorates tissue injury without apparent increase in susceptibility to infection. J Clin Investig. 2005;115:2444–53.
Banda NK, Hyatt S, Antonioli AH, White JT, Glogowska M, Takahashi K, Merkel TJ, Stahl GL, Mueller-Ortiz S, Wetsel R, et al. Role of C3a receptors, C5a receptors, and complement protein C6 deficiency in collagen antibody-induced arthritis in mice. J Immunol. 2012;188:1469–78.
Chowdhury P, Zhou W, Sacks SH. Complement in renal transplantation. Nephron Clin Pract. 2003;95:c3–8.
Colvin RB, Smith RN. Antibody-mediated organ-allograft rejection. Nat Rev Immunol. 2005;5:807–17.
Gu H, Fisher AJ, Mickler EA, Duerson F 3rd, Cummings OW, Peters-Golden M, Twigg HL 3rd, Woodruff TM, Wilkes DS, Vittal R. Contribution of the anaphylatoxin receptors, C3aR and C5aR, to the pathogenesis of pulmonary fibrosis. FASEB J. 2016;30:2336–50.
Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD, Chen Y, Zhang K, Ambati BK, Baffi JZ, Ambati J. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci USA. 2006;103:2328–33.
Riedemann NC, Ward PA. Complement in ischemia reperfusion injury. Am J Pathol. 2003;162:363–7.
Sacks SH, Zhou W, Andrews PA, Hartley B. Endogenous complement C3 synthesis in immune complex nephritis. Lancet. 1993;342:1273–4.
Sacks SH, Zhou W, Pani A, Campbell RD, Martin J. Complement C3 gene expression and regulation in human glomerular epithelial cells. Immunology. 1993;79:348–54.
Stimler NP, Hugli TE, Bloor CM. Pulmonary injury induced by C3a and C5a anaphylatoxins. Am J Pathol. 1980;100:327–48.
Zhou W, Medof ME, Heeger PS, Sacks S. Graft-derived complement as a mediator of transplant injury. Curr Opin Immunol. 2007;19:569–76.
McGregor CG, Ricci D, Miyagi N, Stalboerger PG, Du Z, Oehler EA, Tazelaar HD, Byrne GW. Human CD55 expression blocks hyperacute rejection and restricts complement activation in Gal knockout cardiac xenografts. Transplantation. 2012;93:686–92.
Yamada K, Miwa T, Liu J, Nangaku M, Song W-C. Critical protection from renal ischemia reperfusion injury by CD55 and CD59. J Immunol. 2004;172:3869–75.
Tan CD, Sokos GG, Pidwell DJ, Smedira NG, Gonzalez-Stawinski GV, Taylor DO, Starling RC, Rodriguez ER. Correlation of donor-specific antibodies, complement and its regulators with graft dysfunction in cardiac antibody-mediated rejection. Am J Transplant. 2009;9:2075–84.
Acknowledgements
We gratefully acknowledge the Animal services staff and R. C. logistics and purchasing office for prioritizing all research plans in a timely fashion.
Funding
This research study was approved, and funded under the RAC 215 0017 to MAK (PI) by Office of Research Administration at the King Faisal Specialist Hospital & Research Centre (KFSH&RC), Riyadh, Kingdom of Saudi Arabia.
Author information
Authors and Affiliations
Contributions
All major experiments data analysis and manuscript writing were performed by MAK. TS, SK, AA, and HAA contributed to immunostaining, PCR, flow cytometry and histology experiments, while AMA and DCB provided key suggestions during the whole study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All material is original and has not been reproduced from another source, and experiments using animals were approved by the KFSHRC animal care and use committee.
Consent for publication
As per journal rules, I disclose that (1) All authors agree with the submission; (2) The work has not been published or submitted for publication elsewhere, either completely or in part, or in another form or language.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Khan, M.A., Shamma, T., Kazmi, S. et al. Hypoxia-induced complement dysregulation is associated with microvascular impairments in mouse tracheal transplants. J Transl Med 18, 147 (2020). https://doi.org/10.1186/s12967-020-02305-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-020-02305-z