
Abstract
Background
Lung adenocarcinoma (LUAD) is the most frequent subtype of lung cancer. The prognostic signature could be reliable to stratify LUAD patients according to risk, which helps the management of the systematic treatments. In this study, a systematic and reliable immune signature was performed to estimate the prognostic stratification in LUAD.
Methods
The profiles of immune-related genes for patients with LUAD were used as one TCGA training set: n = 494, other validation set 1: n = 226 and validation set 2: n = 398. Univariate Cox survival analysis was used to identify the candidate immune-related genes from each cohort. Then, the immune signature was developed and validated in the training and validation sets.
Results
In this study, functional analysis showed that immune-related genes involved in immune regulation and MAPK signaling pathway. A prognostic signature based on 10 immune-related genes was established in the training set and patients were divided into high-risk and low-risk groups. Our 10 immune-related gene signature was significantly related to worse survival, especially during early-stage tumors. Further stratification analyses revealed that this 10 immune-related gene signature was still an effective tool for predicting prognosis in smoking or nonsmoking patients, patients with KRAS mutation or KRAS wild-type, and patients with EGFR mutation or EGFR wild-type. Our signature was negatively correlated with B cell, CD4+ T cell, CD8+ T cell, neutrophil, dendritic cell (DC), and macrophage immune infiltration, and immune checkpoint molecules PD-1 and CTLA-4 (P < 0.05).
Conclusions
These findings suggested that our signature was a promising biomarker for prognosis prediction and can facilitate the management of immunotherapy in LUAD.
Similar content being viewed by others


Background
Lung carcinoma is the most frequent human malignant neoplasm and the highest cause of cancer-related deaths worldwide [1]. Despite recent advances in surgery, chemotherapy, radiotherapy, targeted therapy and immunotherapy, the 5-year survival rate of patients with non-small cell lung cancer (NSCLC) remains poor [2, 3]. NSCLC, accounting for approximately 85% of lung cancer cases, consists of two common histological subtypes: lung adenocarcinoma (LUAD) and squamous cell carcinoma (LUSC) [4]. Increasing evidence suggests the well-known biological diversity of LUAD and LUSC. For example, epidermal growth factor receptor (EGFR) mutations are more frequent in LUAD than in LUSC [5]. LUAD is the most common subtype of NSCLC [6].
The immune system has been reported to play an essential role in the development and progression of cancer [7, 8]. Tumor immunotherapy as an important driver of personalized medicine harness the anti-tumor effects of the immune system to obtain a durable cure with minimal toxicity [9, 10]. In recent years, the immune checkpoint proteins such as cytotoxic T-lymphocyte antigen 4 (CTLA-4) or the programmed cell death ligand 1/protein 1 pathway (PD-L1/PD-1) have been used as crucial targets for immunotherapy in many cancers, including LUAD [11, 12]. For example, PD-L1 expression is a predictive biomarker for worse prognosis of NSCLC and the probability of clinical benefit from immune-modulating drugs is greater in NSCLC patients expressing PD-L1 [13, 14]. Most studies on immune-related genes such as PD-L1, CD8A, and CD4 expression have been conducted in NSCLC [15,16,17]. Because the biological differences among the various histological subtypes might have a different therapeutic influence. Moreover, the molecular characteristics describing tumor-immune effects still remain largely unclear in LUAD. Therefore, we focused exclusively on the study of LUAD to further explore the prognostic and predictive significance of the immune-related genes.
In our present work, multiple cohorts were utilized to construct and validate a novel immune prognostic signature for the stratification of LUAD. This study provided more in-depth insight into the prognostic stratification of patients with LUAD as well as provided a tumor-immune interaction with great promise for the therapeutic interventions of LUAD.
Materials and methods
Study samples
The raw expression data (Workflow Type: HTSeq-Counts) for LUAD were enrolled from Genomic Data Commons Data Portal, which fulfilled the approval of the project by the consortium. The Cancer Genome Atlas (TCGA) data were normalized by the Trimmed Mean of M-values (TMM) method and the mean expression levels with ≤ 1 were excluded. Then, the expression data were transformed with log2. For the prognostic information, patients with incomplete follow-up time were excluded. Finally, 494 cases with LUAD had sufficient survival data recorded. The Gene Expression Omnibus (GEO) datasets by microarray were used in the patients with LUAD. IRON normalization was performed on the GSE72094, with log2 expression. The GSE31210 data were normalized by the MAS5 algorithm and the expression data were transformed with log2. The cases with insufficient follow-up time were excluded. Finally, 226 patients from GSE31210 (validation set 1) and 398 cases from GSE72094 (validation set 2) were included.
In this study, TCGA data was applied as a training set. The remaining two cohorts were applied as two validation sets. Overall survival (OS) was estimated from the date of the study enrollment to the recorded date of death of any cause or the last follow-up time. The clinicopathological characteristics of LUAD patients from the training and validation sets are listed in Table 1.
The functional analysis of immune genes
We applied the Immunology Database and Analysis Portal (ImmPort) database to select immune-related genes in immunology research [18, 19]. Using univariate Cox regression analysis, 299 survival-related genes with P < 0.05 were found in the TCGA training set (Additional file 1: Table S1), where further analyzed for functional enrichment analysis. The functional analysis was conducted using clusterProfiler package [20] for gene ontology (GO) terms and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.
Development of the immune-related gene prognostic signature
Immune-related genes were obtained from the ImmPort database (https://immport.niaid.nih.gov) (Additional file 2: Table S2). We performed the following process to establish the immune signature. First, univariate Cox regression analysis with P < 0.05 was conducted to find the potential immune-related prognostic genes. Three data sets were used to get overlapping genes, which can increase the credibility of the potential immune-related prognostic genes, the final 52 overlapping survival-related immune genes were found (Additional file 3: Table S3 and Additional file 4: Figure S1). Second, to achieve the final immune-related prognostic genes in our model, multivariate Cox regression analysis based on the two-step method was finally carried out for these 52 candidate immune-related genes in the TCGA training set. Finally, 10 immune-related genes were identified from the TCGA cohort in the present model (Additional file 5: Table S4).
Tumor-infiltrating immune cells
TIMER was applied to estimate the abundance of tumor-infiltrating immune cells in the tumor microenvironment (TME) (http://cistrome.dfci.harvard.edu/TIMER/), including six immune cell types: B cell, CD4 T cell, CD8 T cell, neutrophil, macrophage, and dendritic cell (DC). We investigated whether our signature played a role in immune infiltration.
Statistical analysis
Spearman’s correlation coefficient was used to assess the association between risk score and tumor-infiltrating immune cells and immune checkpoint molecules. The cut-off value was determined based on the median value in the training set. Patients were divided into low-risk group and high-risk group. To examine significant differences of between the low-risk group and the corresponding high-risk group, Kaplan–Meier survival methods with the log-rank test were applied. To estimate the predictive accuracy of the immune-related gene signature, time-dependent receiver-operating characteristic (ROC) method was conducted in all datasets. Univariate and multivariate survival analyses were conducted using the Cox proportional hazard model to determine whether there was a significant relationship between the immune signature and the survival of patients with LUAD, along with the clinicopathological variables in the training and validation sets. Subgroup analyses were also carried out according to the stratification of age, gender, smoking status, tumor stage, KRAS mutation, and EGFR mutation status. All data were analyzed by using R software (version 3.5.1; R Foundation for Statistical Computing, Vienna, Austria).
Results
The molecular mechanism of immune-related genes
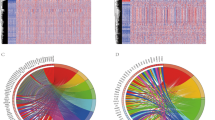
299 survival-related immune genes were performed for gene enrichment analysis. As shown in Fig. 1, KEGG analysis demonstrated that MAPK, Ras, Rap1, ErbB pathways, EGFR tyrosine kinase inhibitor resistance, proteasome, and endocrine resistance were associated with these immune genes (Fig. 1a). GO analysis showed that the immune-related mechanisms such as immune response-activating/regulating cell surface receptor signaling pathway, T cell activation, T cell receptor signaling pathway, antigen receptor-mediated signaling pathway, antigen processing and presentation, cytokine activity, and MHC protein complex etc. were enriched (Fig. 1b).

Functional enrichment analysis of immune-related genes in LUAD. a Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. b Gene ontology (GO) analysis on biological process (BP), cellular component (CC), and molecular function (MF)
Construction of a 10 immune-related gene signature
We finally applied the multivariate Cox regression model to select the final genes in the training cohort. Eventually, 10 immune-related genes were used to construct a prognostic model in LUAD. A prognostic index was established based on the expression level and the corresponding regression coefficient of each immune-related gene, the following formula was present: [FURIN* 0.1089] + [PSMD14*(− 0.2344)], [ARRB1*(− 0.2738)] + (TUBB3*0.1028) + (ADM*0.1159) + [ZAP70*(− 0.2977)] + [RFXAP*(− 0.2549)] + [SHC3*(− 0.1522)] +[BMP5*(− 0.0763)] + (CD40LG*0.1625).
A 10 immune-related gene signature predicts survival in LUAD
The distribution and survival status of patients for the 10 immune-related gene signature were shown in the training and validation sets (Fig. 2a). Time-dependent ROC results were used to evaluate the predictive capacity of a 10 immune-related gene signature at 1, 3, and 5 years (Fig. 2b). The AUCs (Area under the ROC curve) for the long-time survival at 5 years were > 0.70 in the training set and the other two validation cohorts, indicating a good accuracy of the 10 immune-related gene signature for survival prediction. Kaplan–Meier curves showed that patients with LUAD in the high-risk group had closely shorter prognosis than patients with LUAD in the low-risk group (all P values < 0.001) (Fig. 2c).

The prognostic signature based on 10 immune-related genes in LUAD. a The distribution and survival status of patients for the immune-related gene signature. b Time-dependent ROC results at 1, 3, and 5 years. c Kaplan–Meier curves between the high-risk and low-risk groups
Independent prognostic and predictive value of the 10 immune-related gene signature
To examine whether our 10 immune-related gene signature was an independent molecular factor for survival prediction in the training and validation sets, univariate and multivariate Cox models were further carried out in this study (Table 2). Univariate Cox analysis showed that the high-risk group was closely associated with worse survival of LUAD in TCGA training set (494 cases: HR = 2.57, 95% CI 1.85–3.56, P < 0.001), validation set 1 (226 cases: HR = 9.11, 95% CI 3.21–25.82, P < 0.001), and validation set 2 (398 cases: HR = 2.80, 95% CI 1.87–4.17, P < 0.001). After adjusting for clinical and pathologic factors, further multivariate Cox analysis suggested that our immune signature was still a novel independent molecular indicator for predicting worse survival in LUAD in TCGA training set (HR = 1.68, 95% CI 1.11–2.54, P = 0.013), validation set 1 (HR = 8.63, 95% CI 2.95–25.21, P < 0.001), and validation set 2 (HR = 2.38, 95% CI 1.57–3.62, P < 0.001).
Predictive role of the 10 immune-related gene signature with the survival in various clinical and mutational characteristics
Stratification analyses were conducted based on age (≥ 65 vs. < 65 years), gender (male and female), smoking behavior (smoking and nonsmoking), tumor stage (stage 3–4: advanced-stage and stage 1–2: early-stage), KRAS mutation status (mutation and wild-type) and EGFR mutation status (mutation and wild-type) in the entire set (Figs. 3, 4). The cut-off value was 0.9325 and cases were divided into high- and low-risk groups. Because patients with various T, M, N, and TP53 mutation status were conducted in only a cohort, these clinical variables such as T, M, N, and TP53 mutation status were removed from subgroup analyses. For early-stage patients, the high-risk group indicated closely poor prognosis than the low-risk group (P < 0.05), but no significant prognostic relationship was observed between the high- and low-risk groups for advanced-stage patients (Fig. 4), which might cause owing to the small study population of advanced-stage patients. The results demonstrated that high-risk LUAD patients in each stratum of age, gender, smoking behavior, KRAS mutation, and EGFR mutation status presented worse survival than low-risk LUAD patients (all P values < 0.05) (Figs. 3, 4), suggesting that our 10 immune-related gene signature-based risk group stratification was still an effective tool for survival prediction in older or younger, male or female, and smoking or nonsmoking patients with LUAD, patients with KRAS mutation or KRAS wild-type, and patients with EGFR mutation or EGFR wild-type.

Kaplan-Meier analyses of LUAD patients with age, gender, and smoking behavior, including a ≥ 65 years, b < 65 years, c male, d female, e smoking, and f nonsmoking

Kaplan–Meier analyses of LUAD patients with stage, KRAS mutation status, and EGFR mutation status, including a stage 3–4, b stage 1–2, c KRAS mutation, d KRAS wild-type, e EGFR mutation, and f EGFR wild-type
A stratification analysis of stage 1 and stage 2 tumors was also performed. The result showed that the high-risk group was closely correlated with poor prognosis in stage 1 and stage 2 LUAD tumors (P < 0.05) (Additional file 6: Figure S2), suggesting that our immune signature-based risk group stratification was still an effective tool for prognosis prediction in stage 1 and stage 2 tumors. Moreover, multivariate Cox analysis within early stages of LUAD tumors demonstrated that our immune signature was still an independent molecular factor for predicting survival (HR = 3.93, 95% CI 2.38–6.5, P < 0.0001) (Additional file 7: Table S5).
Tumor-infiltrating immune cells
We analyzed whether our immune-related gene signature was related to immune infiltration in LUAD, such as B cell, CD4 T cell, CD8 T cell, neutrophil, macrophage, and dendritic cell (DC). As shown in Fig. 5, our immune-related gene signature was negatively correlated with B cells (r = − 0.40, P < 0.001), CD4+ T cells (r = − 0.27, P < 0.001), DCs (r = − 0.22, P < 0.001), CD8+ T cells (r = − 0.15, P = 0.001), neutrophils (r = − 0.12, P = 0.011), and macrophages (r = − 0.11, P = 0.012).

Correlation between our signature and tumor-infiltrating immune cells. a Association between risk score and B cells. b Association between risk score and CD4+ T cells. c Association between risk score and CD8+ T cells. d Association between risk score and Neutrophils. e Association between risk score and dendritic cells (DCs). f Association between risk score and Macrophages
Association between our signature and immune checkpoint molecules
Immune checkpoint blockade (ICB) therapy such as immune-checkpoint proteins programmed cell death 1 (PD-1), programmed cell death-ligand 1 (PD-L1), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) targets has been reported to provide clinical benefits for cancer immunotherapy in some advanced-cancers such as melanoma and NSCLC [21, 22]. We investigated the correlation between our signature and the immune checkpoint molecules PD-1, PD-L1, PD-L2, and CTLA-4 in LUAD. The results showed that our signature was negatively associated with PD-1 (r = − 0.11, P = 0.017) and CTLA-4 (r = − 0.25, P < 0.001) (Fig. 6a, b). In addition, PD-1, PD-L1, PD-L2, and CTLA-4 were found to be coexpressed in LUAD (P < 0.001) (Fig. 6C).

Correlation between our signature and known immune checkpoint genes. a Association between risk score and PD-1. b Association between risk score and CTLA-4. c Association of each immune checkpoint gene. “x” for no correlation; blue for negative correlation; red for positive correlation
Discussion
The most frequent histological subtypes of lung cancer are LUAD and frequently occurs in females and nonsmoking people [23]. LUAD is related to distinct oncogene alterations. The common somatic gene aberrations are EGFR and KRAS mutations, and ALK rearrangements, which have been extensively reported and studied in LUAD [24, 25]. The frequency of oncogenic driver aberrations is different based on ethnicity, gender, and smoking behavior, which may result in differences in treatment efficacy [26]. In addition, in recent years reliable prognostic biomarkers have already appeared that are modifying the prognosis, which are applied to select groups of patients who are at high-risk and who may benefit from the personalized therapy [27,28,29], but their accuracy of survival estimation is not shown and remains limited. Therefore, it is imperative to identify precise biomarkers of LUAD and to select the appropriate immunotherapy for improving survival of this disease. In our current work, we constructed and validated a new prognostic signature based on 10 immune-related genes for survival prediction in patients with LUAD.
The immune system has a key role in cancer development progression [7, 8]. Such as, MAPK signaling involves in cell proliferation, apoptosis and immune escape, which contributes to the progression of the tumor [30]. Cytokines regulate tumor growth, survival, and invasion, and metastatic colonization [31]. LUAD shows altered T cell and NK cell compartments [32]. Choi et al. reported that high ImmuneScore was associated with favorable prognosis in LUAD [33]. However, immune-related molecular mechanisms involved in LUAD remain largely unclear. In this study, immune-related genes were found to be associated with immune regulation (i.e. immune response-activating/regulating cell surface receptor signaling pathway, T cell activation, and cytokine activity etc.) and biological pathways such as MAPK signaling in LUAD. Further 10 immune-related gene signature for survival prediction was identified in LUAD. Immune signatures can predict prognosis across solid tumors [34]. For example, a proposed clinical-immune signature is significantly correlated with worse prognosis and can be a promising biomarker for predicting overall survival in ovarian cancer [35]. The seven immune-related gene signature is significantly related to poor survival and can serve as a potential marker for reflecting the prognosis in clear renal clear cell carcinoma [36]. A prognostic immune signature can predict cervical cancer patients’ survival [37]. The nine immune-related gene signature is correlated with worse prognosis and is a potential predictive biomarker in hepatocellular carcinoma [38]. The previous findings were consistent with our current result that the high-risk patients were related to short survival time and worse survival of LUAD. Then, multivariate Cox analysis indicated that our immune signature remained an independent molecular indicator for survival prediction. These analyses confirmed that our immune signature was an effective and good performance for predicting prognosis in LUAD, which was reasonable and reliable.
This model for LUAD contained 10 immune-related genes. Among them, FURIN correlates with many cancer-related processes such as cell proliferation, migration, invasion, and angiogenesis, which promotes tumor progression [39]. FURIN has also a crucial function in the adaptive immunity [40]. PSMD14 induces cell cycle and senescence and may participate in the role of the proteasome [41, 42]. ARRB1 mediates signaling pathways and correlates with the regulation of DNA damage response [43]. ARRB1 participates in cell invasion and proliferation, thereby contributing to NSCLC progression [44]. TUBB3 is a key mechanism of drug resistance and TUBB3 expression shows predictive value for the prognosis in many cancers [45]. SHC3 regulates signal transduction, thereby stimulating hepatocellular carcinoma proliferation, migration, invasion, and epithelial-to-mesenchymal transition (EMT) [46]. BMP5 activates multiple signaling pathways such as p38 MAPK signaling [47]. CD40LG polymorphism is related to various immunological disorders such as tumors [48]. Adrenomedullin (ADM) correlates with cellular growth, anti-apoptotic property, angiogenesis, tumor cell motility and metastasis, inflammatory and immune responses. And ADM is also a significant factor for worse survival in some cancers [49, 50]. ZAP70 is related to the T cell antigen receptor (TCR) complex and is required for T cell activation [51, 52]. RFXAP is a key transcription factor for major histocompatibility complex (MHC) II [53]. On the basis of the above findings, our work integrated 10 immune-related genes into a single panel, found patients in the high immune-risk group associated with worse survival, and confirmed the predictive value of our signature in LUAD. The high abundance of T cell and B-cell is correlated with improved survival in many cancer types, including lung cancer [54]. A high level of tumor-infiltrating lymphocytes is associated with better prognosis of LUAD [55]. High B-cell and CD8+ T cell infiltration is reported to be associated with favorable prognosis in LUAD [54, 56]. The immune checkpoint molecules PD-1 and CTLA-4 treatment is a promising cancer immunotherapy approach for clinical benefits [21]. Studies have reported the coexpression PD-1, PD-L1, PD-L2, and CTLA-4 immune checkpoint molecules [57]. In our study, we also found PD-1, PD-L1, PD-L2, and CTLA-4 immune checkpoint molecules were coexpressed in LUAD. Although our signature was negatively correlated with tumor-infiltrating immune cells and PD-1 and CTLA-4, the correlation was not very significant. Biomarkers of response to ICB do not frequently only focus on the target molecules, but rather inflammatory/interferon signaling cascades [58,59,60]. Thus, our signature may not have a significant response to immune checkpoint molecules. Additional data on the association of our model with other signatures (either a cytolytic signature, IFNG signature, chemokine signature, or the tumor inflammation signature) could be needed in the future.
The common somatic mutations are EGFR and KRAS in LUAD [24]. Hsiao et al. reported that EGFR mutations were closely correlated with therapeutic efficacy and progression-free survival in LUAD [61]. We further evaluated the predictive effect of our 10 immune-related gene signature in different clinical and molecular features. We found that our 10 immune-related gene signature remained a strongly powerful tool for predicting prognosis in older or younger, male or female, and smoking or nonsmoking patients with LUAD, patients with KRAS mutation or KRAS wild-type, and patients with EGFR mutation or EGFR wild-type. Our immune signature in the high-risk group was observed to be associated with worse prognosis for early-stage tumors. Further multivariate Cox analysis showed that our immune signature was still an independent molecular indicator for survival prediction in early-stage LUAD tumors.
These are several limitations in this work that should be noted. First, our study was a retrospective design, more prospective clinical data sets as possible are needed to validate our result in the future. Second, our immune signature was developed by numerous genes, further biological functions are warranted to be further explored in LUAD. Third, our immune signature was calculated based on the gene expression values. Thus, intra-tumor heterogeneity supported by genetic and phenomenological data, which could cause sampling bias.
Conclusion
In conclusion, a 10 immune-related gene signature was built for prognosis prediction in LUAD. This study demonstrated that this 10 immune-related gene signature may become a promising prognostic marker for LUAD, especially in early-stage patients, which could facilitate the personalized therapy and serve new immunotherapy of LUAD. Further studies are still required to prove this signature in LUAD in the future.
Data availability statement
The data sets used are available from the corresponding author on a reasonable request.
Abbreviations
- LUAD:
-
Lung adenocarcinoma
- NSCLC:
-
Non-small cell lung cancer
- LUSC:
-
Lung squamous cell carcinoma
- EGFR:
-
Epidermal growth factor receptor
- TCGA:
-
The Cancer Genome Atlas
- TMM:
-
Trimmed Mean of M-values
- GEO:
-
The Gene Expression Omnibus
- OS:
-
Overall survival
- ImmPort:
-
The Immunology Database and Analysis Portal
- GO:
-
Gene ontology
- KEGG:
-
The Kyoto Encyclopedia of Genes and Genomes
- ROC:
-
Receiver-operating characteristic
- ADM:
-
Adrenomedullin
- EMT:
-
Epithelial-to-mesenchymal transition
- MHC:
-
Major histocompatibility complex
- DC:
-
Dendritic cell
- TME:
-
Tumor microenvironment
- ICB:
-
Immune checkpoint blockade
- PD-1:
-
Programmed cell death 1
- PD-L1:
-
Programmed cell death-ligand 1
- PD-L2:
-
Programmed cell death-ligand 2
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Li L, Sun Y, Feng M, Wang L, Liu J. Clinical significance of blood-based miRNAs as biomarkers of non-small cell lung cancer. Oncol Lett. 2018;15:8915–25.
Ettinger DS, Wood DE, Aisner DL, Akerley W, Bauman J, Chirieac LR, D’Amico TA, DeCamp MM, Dilling TJ, Dobelbower M, et al. Non-small cell lung cancer, version 5.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2017;15:504–35.
Ni S, Ye M, Huang T. Short stature homeobox 2 methylation as a potential noninvasive biomarker in bronchial aspirates for lung cancer diagnosis. Oncotarget. 2017;8:61253–63.
Molina-Pinelo S, Gutierrez G, Pastor MD, Hergueta M, Moreno-Bueno G, Garcia-Carbonero R, Nogal A, Suarez R, Salinas A, Pozo-Rodriguez F, et al. MicroRNA-dependent regulation of transcription in non-small cell lung cancer. PLoS ONE. 2014;9:e90524.
Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–20.
Ferris RL. Immunology and immunotherapy of head and neck cancer. J Clin Oncol. 2015;33:3293–304.
Wormann SM, Diakopoulos KN, Lesina M, Algul H. The immune network in pancreatic cancer development and progression. Oncogene. 2014;33:2956–67.
Koo SL, Wang WW, Toh HC. Cancer immunotherapy—the target is precisely on the cancer and also not. Ann Acad Med Singapore. 2018;47:381–7.
Helmy KY, Patel SA, Nahas GR, Rameshwar P. Cancer immunotherapy: accomplishments to date and future promise. Ther Deliv. 2013;4:1307–20.
Chalela R, Curull V, Enriquez C, Pijuan L, Bellosillo B, Gea J. Lung adenocarcinoma: from molecular basis to genome-guided therapy and immunotherapy. J Thorac Dis. 2017;9:2142–58.
Teglasi V, Reiniger L, Fabian K, Pipek O, Csala I, Bago AG, Varallyai P, Vizkeleti L, Rojko L, Timar J, et al. Evaluating the significance of density, localization, and PD-1/PD-L1 immunopositivity of mononuclear cells in the clinical course of lung adenocarcinoma patients with brain metastasis. Neuro Oncol. 2017;19:1058–67.
Thakur MK, Gadgeel SM. Predictive and prognostic biomarkers in non-small cell lung cancer. Semin Respir Crit Care Med. 2016;37:760–70.
Wang A, Wang HY, Liu Y, Zhao MC, Zhang HJ, Lu ZY, Fang YC, Chen XF, Liu GT. The prognostic value of PD-L1 expression for non-small cell lung cancer patients: a meta-analysis. Eur J Surg Oncol. 2015;41:450–6.
Li B, Cui Y, Diehn M, Li R. Development and validation of an individualized immune prognostic signature in early-stage nonsquamous non-small cell lung cancer. JAMA Oncol. 2017;3:1529–37.
Prat A, Navarro A, Pare L, Reguart N, Galvan P, Pascual T, Martinez A, Nuciforo P, Comerma L, Alos L, et al. Immune-Related gene expression profiling after PD-1 blockade in non-small cell lung carcinoma, head and neck squamous cell carcinoma, and melanoma. Cancer Res. 2017;77:3540–50.
Bouillez A, Rajabi H, Jin C, Samur M, Tagde A, Alam M, Hiraki M, Maeda T, Hu X, Adeegbe D, et al. MUC1-C integrates PD-L1 induction with repression of immune effectors in non-small-cell lung cancer. Oncogene. 2017;36:4037–46.
Bhattacharya S, Dunn P, Thomas CG, Smith B, Schaefer H, Chen J, Hu Z, Zalocusky KA, Shankar RD, Shen-Orr SS, et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci Data. 2018;5:180015.
Bhattacharya S, Andorf S, Gomes L, Dunn P, Schaefer H, Pontius J, Berger P, Desborough V, Smith T, Campbell J, et al. ImmPort: disseminating data to the public for the future of immunology. Immunol Res. 2014;58:234–9.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7.
Nishino M, Ramaiya NH, Hatabu H, Hodi FS. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol. 2017;14:655–68.
Champiat S, Lambotte O, Barreau E, Belkhir R, Berdelou A, Carbonnel F, Cauquil C, Chanson P, Collins M, Durrbach A, et al. Management of immune checkpoint blockade dysimmune toxicities: a collaborative position paper. Ann Oncol. 2016;27:559–74.
Saito M, Shiraishi K, Kunitoh H, Takenoshita S, Yokota J, Kohno T. Gene aberrations for precision medicine against lung adenocarcinoma. Cancer Sci. 2016;107:713–20.
Calvayrac O, Pradines A, Pons E, Mazieres J, Guibert N. Molecular biomarkers for lung adenocarcinoma. Eur Respir J. 2017;49:1601734.
Devarakonda S, Morgensztern D, Govindan R. Genomic alterations in lung adenocarcinoma. Lancet Oncol. 2015;16:e342–51.
Saito M, Suzuki H, Kono K, Takenoshita S, Kohno T. Treatment of lung adenocarcinoma by molecular-targeted therapy and immunotherapy. Surg Today. 2018;48:1–8.
Shukla S, Evans JR, Malik R, Feng FY, Dhanasekaran SM, Cao X, Chen G, Beer DG, Jiang H, Chinnaiyan AM. Development of a RNA-Seq based prognostic signature in lung adenocarcinoma. J Natl Cancer Inst. 2017;109:1–9.
Wistuba II, Behrens C, Lombardi F, Wagner S, Fujimoto J, Raso MG, Spaggiari L, Galetta D, Riley R, Hughes E, et al. Validation of a proliferation-based expression signature as prognostic marker in early stage lung adenocarcinoma. Clin Cancer Res. 2013;19:6261–71.
Director’s Challenge Consortium for the Molecular Classification of Lung A, Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, Gerald WL, Eschrich S, Jurisica I, Giordano TJ, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med. 2008;14:822–7.
Peluso I, Yarla NS, Ambra R, Pastore G, Perry G. MAPK signalling pathway in cancers: Olive products as cancer preventive and therapeutic agents. Semin Cancer Biol. 2019;56:185–95.
Yao M, Brummer G, Acevedo D, Cheng N. Cytokine regulation of metastasis and tumorigenicity. Adv Cancer Res. 2016;132:265–367.
Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell. 2017;169(750–765):e717.
Choi H, Na KJ. Integrative analysis of imaging and transcriptomic data of the immune landscape associated with tumor metabolism in lung adenocarcinoma: clinical and prognostic implications. Theranostics. 2018;8:1956–65.
Hsu DS, Kim MK, Balakumaran BS, Acharya CR, Anders CK, Clay T, Lyerly HK, Drake CG, Morse MA, Febbo PG. Immune signatures predict prognosis in localized cancer. Cancer Investig. 2010;28:765–73.
Shen S, Wang G, Zhang R, Zhao Y, Yu H, Wei Y, Chen F. Development and validation of an immune gene-set based prognostic signature in ovarian cancer. EBioMedicine. 2019;40:318–26.
Shen C, Liu J, Wang J, Zhong X, Dong D, Yang X, Wang Y. Development and validation of a prognostic immune-associated gene signature in clear cell renal cell carcinoma. Int Immunopharmacol. 2020;81:106274.
Yang S, Wu Y, Deng Y, Zhou L, Yang P, Zheng Y, Zhang D, Zhai Z, Li N, Hao Q, et al. Identification of a prognostic immune signature for cervical cancer to predict survival and response to immune checkpoint inhibitors. Oncoimmunology. 2019;8:e1659094.
Wang Z, Zhu J, Liu Y, Liu C, Wang W, Chen F, Ma L. Development and validation of a novel immune-related prognostic model in hepatocellular carcinoma. J Transl Med. 2020;18:67.
Jaaks P, Bernasconi M. The proprotein convertase furin in tumour progression. Int J Cancer. 2017;141:654–63.
Vahatupa M, Aittomaki S, Martinez Cordova Z, May U, Prince S, Uusitalo-Jarvinen H, Jarvinen TA, Pesu M. T-cell-expressed proprotein convertase FURIN inhibits DMBA/TPA-induced skin cancer development. Oncoimmunology. 2016;5:e1245266.
Zhang L, Hao C, Li J, Qu Y, Bao L, Li Y, Yue Z, Zhang M, Yu X, Chen H, et al. Bioinformatics methods for identifying differentially expressed genes and signaling pathways in nano-silica stimulated macrophages. Tumour Biol. 2017;39:1010428317709284.
Byrne A, McLaren RP, Mason P, Chai L, Dufault MR, Huang Y, Liang B, Gans JD, Zhang M, Carter K, et al. Knockdown of human deubiquitinase PSMD14 induces cell cycle arrest and senescence. Exp Cell Res. 2010;316:258–71.
Shen H, Wang L, Zhang J, Dong W, Zhang T, Ni Y, Cao H, Wang K, Li Y, Wang Y, Du J. ARRB1 enhances the chemosensitivity of lung cancer through the mediation of DNA damage response. Oncol Rep. 2017;37:761–7.
Dasgupta P, Rizwani W, Pillai S, Davis R, Banerjee S, Hug K, Lloyd M, Coppola D, Haura E, Chellappan SP. ARRB1-mediated regulation of E2F target genes in nicotine-induced growth of lung tumors. J Natl Cancer Inst. 2011;103:317–33.
Person F, Wilczak W, Hube-Magg C, Burdelski C, Moller-Koop C, Simon R, Noriega M, Sauter G, Steurer S, Burdak-Rothkamm S, Jacobsen F. Prevalence of betaIII-tubulin (TUBB3) expression in human normal tissues and cancers. Tumour Biol. 2017;39:1010428317712166.
Liu Y, Zhang X, Yang B, Zhuang H, Guo H, Wei W, Li Y, Chen R, Li Y, Zhang N. Demethylation-induced overexpression of Shc3 drives c-Raf-Independent activation of MEK/ERK in HCC. Cancer Res. 2018;78:2219–32.
Snelling SJ, Hulley PA, Loughlin J. BMP5 activates multiple signaling pathways and promotes chondrogenic differentiation in the ATDC5 growth plate model. Growth Factors. 2010;28:268–79.
Aloui C, Sut C, Cognasse F, Granados V, Hassine M, Chakroun T, Garraud O, Laradi S. Development of a highly resolutive method, using a double quadruplex tetra-primer-ARMS-PCR coupled with capillary electrophoresis to study CD40LG polymorphisms. Mol Cell Probes. 2015;29:335–42.
Ferrero H, Larrayoz IM, Gil-Bea FJ, Martinez A, Ramirez MJ. Adrenomedullin, a novel target for neurodegenerative diseases. Mol Neurobiol. 2018;55:8799–814.
Larrayoz IM, Martinez-Herrero S, Garcia-Sanmartin J, Ochoa-Callejero L, Martinez A. Adrenomedullin and tumour microenvironment. J Transl Med. 2014;12:339.
Au-Yeung BB, Shah NH, Shen L, Weiss A. ZAP-70 in signaling, biology, and disease. Annu Rev Immunol. 2018;36:127–56.
Moore JC, Mulligan TS, Yordan NT, Castranova D, Pham VN, Tang Q, Lobbardi R, Anselmo A, Liwski RS, Berman JN, et al. T cell immune deficiency in zap70 mutant zebrafish. Mol Cell Biol. 2016;36:2868–76.
Long AB, Ferguson AM, Majumder P, Nagarajan UM, Boss JM. Conserved residues of the bare lymphocyte syndrome transcription factor RFXAP determine coordinate MHC class II expression. Mol Immunol. 2006;43:395–409.
Iglesia MD, Parker JS, Hoadley KA, Serody JS, Perou CM, Vincent BG. Genomic analysis of immune cell infiltrates across 11 tumor types. J Natl Cancer Inst. 2016;108:1–11.
Kim A, Lee SJ, Ahn J, Park WY, Shin DH, Lee CH, Kwon H, Jeong YJ, Ahn HY, I H, et al. The prognostic significance of tumor-infiltrating lymphocytes assessment with hematoxylin and eosin sections in resected primary lung adenocarcinoma. PLoS ONE. 2019;14:e0224430.
Varn FS, Tafe LJ, Amos CI, Cheng C. Computational immune profiling in lung adenocarcinoma reveals reproducible prognostic associations with implications for immunotherapy. Oncoimmunology. 2018;7:e1431084.
Guo W, Zhu L, Zhu R, Chen Q, Wang Q, Chen JQ. A four-DNA methylation biomarker is a superior predictor of survival of patients with cutaneous melanoma. Elife. 2019;8:e44310.
Di Pilato M, Kim EY, Cadilha BL, Prussmann JN, Nasrallah MN, Seruggia D, Usmani SM, Misale S, Zappulli V, Carrizosa E, et al. Targeting the CBM complex causes Treg cells to prime tumours for immune checkpoint therapy. Nature. 2019;570:112–6.
Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–201.
Lou Y, Diao L, Cuentas ER, Denning WL, Chen L, Fan YH, Byers LA, Wang J, Papadimitrakopoulou VA, Behrens C, et al. Epithelial–mesenchymal transition is associated with a distinct tumor microenvironment including elevation of inflammatory signals and multiple immune checkpoints in lung adenocarcinoma. Clin Cancer Res. 2016;22:3630–42.
Hsiao SH, Lin HC, Chou YT, Lin SE, Kuo CC, Yu MC, Chung CL. Impact of epidermal growth factor receptor mutations on intracranial treatment response and survival after brain metastases in lung adenocarcinoma patients. Lung Cancer. 2013;81:455–61.
Acknowledgements
We gratefully acknowledge Genomic Data Commons Data Portal and Gene Expression Omnibus.
Funding
None.
Author information
Authors and Affiliations
Contributions
DG and JZ were responsible for the conception and designed this work. DG, MW, ZS, and JZ drafted of the article and were responsible for the final approval of the submission. DG, MW, ZS, and JZ analyzed data and were responsible for the interpretation and completion of the figures and tables. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All data collection and processing, including the consenting process, were performed after approval by all local institutional review boards and in accord with Genomic Data Commons Data Portal and Gene Expression Omnibus Human Subjects Protection and Data Access Policies.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
299 survival-related genes for enrichment analysis.
Additional file 2: Table S2.
Immune-related genes from the ImmPort database.
Additional file 3: Table S3.
52 potential survival-related immune genes from three cohorts.
Additional file 4: Figure S1.
Venn diagram of the overlapping immune-related genes with the survival using univariate Cox analysis with P < 0.05 from three data sets.
Additional file 5: Table S4.
Final 10 immune-related genes in the current model.
Additional file 6: Figure S2.
Kaplan–Meier analyses of our immune signature in stage 1 and stage 2 tumors, including (A) stage 1 lung adenocarcinoma (LUAD) and (B) stage 2 LUAD.
Additional file 7: Table S5.
Multivariate Cox analyses of our immune signature in early-stage lung adenocarcinoma (LUAD).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Guo, D., Wang, M., Shen, Z. et al. A new immune signature for survival prediction and immune checkpoint molecules in lung adenocarcinoma. J Transl Med 18, 123 (2020). https://doi.org/10.1186/s12967-020-02286-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-020-02286-z