
Abstract
Background
Helicobacter pylori (H. pylori) is a gram-negative bacterium that chronically infects approximately 50% of the world’s human population. While in most cases the infection remains asymptomatic, 10% of infected individuals develop gastric pathologies and 1–3% progress to gastric cancer. AlthoughH. pylori induces severe inflammatory responses, the host’s immune system fails to clear the pathogen and H. pylori can persist in the human stomach for decades. As suppressor of cytokine signaling (SOCS) proteins are important feedback regulators limiting inflammatory responses, we hypothesized thatH. pylori could modulate the host’s immune responses by inducing SOCS expression.
Methods
The phenotype of human monocyte-derived DCs (moDCs) infected withH. pylori was analyzed by flow cytometry and multiplex technology. SOCS expression levels were monitored by qPCR and signaling studies were conducted by means of Western blot. For functional studies, RNA interference-based silencing of SOCS1–3 and co-cultures with CD4+ T cells were performed.
Results
We show that H. pylori positive gastritis patients express significantly higher SOCS3, but not SOCS1 andSOCS2, levels compared to H. pylori negative patients. Moreover, infection of human moDCs with H. pylori rapidly inducesSOCS3 expression, which requires the type IV secretion system (T4SS), release of TNFα, and signaling via the MAP kinase p38, but appears to be independent of TLR2, TLR4, MEK1/2 and STAT proteins. Silencing of SOCS3 expression in moDCs prior to H. pylori infection resulted in increased release of both pro- and anti-inflammatory cytokines, upregulation of PD-L1, and decreased T-cell proliferation.
Conclusions
This study shows that H. pylori induces SOCS3 via an autocrine loop involving the T4SS and TNFα and p38 signaling. Moreover, we demonstrate that high levels of SOCS3 in DCs dampen PD-L1 expression on DCs, which in turn drives T-cell proliferation.
Video Abstract
Similar content being viewed by others


Background
With more than 4 billion infected persons, Helicobacter pylori is one of the most prevalent human pathogens worldwide. H. pylori infection is characterized by persistent colonization of the gastric mucosa [1] associated with leukocyte infiltration and increased secretion of pro-inflammatory cytokines within the first 2 weeks of infection [2, 3]. Without antibiotic treatment, however, the host’s immune system fails to clear the bacterial burden and H. pylori infection lasts for the entire life of the host [4]. Therefore, infected individuals experience chronic infections which can give rise to severe gastritis, several ulcer entities and gastric cancer [5,6,7]. Accordingly,H. pylori was categorized as a class I (definite) carcinogen by the World Health Organization (WHO) in 1994 [8]. H. pylori harbors several virulence factors, including the vacuolating toxin VacA, the serine protease HtrA, and a pathogenicity island encoding a type IV secretion system (T4SS) which delivers bacterial factors directly into the host cell cytoplasm (cagPAI). These latter factors include the bacterial protein CagA, peptidoglycan, and ADP-glycero-β-D-manno-heptose (ADP heptose) and are thought to hijack host cell signaling networks [9, 10].
In stomach biopsies of H. pylori-infected individuals, increased numbers of tissue-infiltrating immune cells have been identified, including neutrophils [11], B and T cells [12], and dendritic cells (DCs) [13]. Moreover, it has been reported thatH. pylori infection results in recruitment of myeloid DCs to the inflamed mucosa. In contrast, biopsies from uninfected individuals lack myeloid DCs [14]. Furthermore, DCs were shown to take up virulence products of H. pylori [15] and to play key roles in initiating adaptive immune responses toward H. pylori [16]. However, the situation in H. pylori infections is ambiguous. Despite effective evasion from Toll-like receptor-4- (TLR4) and TLR5-mediated pathogen recognition, significant DC activation is observed [17,18,19]. While the effects of H. pylori infection on epithelial cells have been extensively studied, the consequences for human DCs are less well characterized.
Stimulation of DCs with bacterial components results in DC activation and maturation, which involves a wide variety of signaling cascades and results in the secretion of pro-inflammatory mediators as well as presentation of processed antigen in the context of co-stimulatory molecules. Mature DCs thus provide important signals that determine the development of different pathogen-specific T-helper cell subgroups, which in turn are crucial for protective immunity. A strong inflammatory response ensures killing of pathogens; however, to avoid excessive inflammation, several mechanisms have evolved to tightly regulate these processes. A well-described group of immune regulators are the suppressor of cytokine signaling (SOCS) proteins [20]. The SOCS protein family consists of 8 members (SOCS1–7, CIS) which are upregulated upon stimulation of cytokine receptors and TLRs [21,22,23]. SOCS proteins inhibit immune signaling at different levels of signal transduction, including inhibition of receptor-associated kinases, competition with signal transducers and activators of transcription (STATs), and proteasomal degradation of signaling molecules. Hence, SOCS proteins play an important role in limiting excessive immune responses [21]. However, recent studies indicate that SOCS proteins not only regulate cytokine signaling, but also have a direct impact on the inflammatory potential of DCs [23, 24].
While several studies have described a pro-inflammatory role of DCs inH. pylori infection, other data suggest thatH. pylori-infected DCs are more likely to have a regulatory phenotype [16]. Since SOCS proteins can shift the balance between the pro- and anti-inflammatory potential of DCs [23,24,25,26,27,28], and because the role of SOCS proteins during H. pylori infection has been underexplored, this study aimed to shed light on SOCS proteins as possible regulators of DC function in response to H. pylori infection. Here, we show that human DCs rapidly upregulateSOCS3 expression after infection with wild-type H. pylori P12, whereas the induction ofSOCS1 and SOCS2 expression is observed only at later time points. ImmediateH. pylori-induced SOCS3 expression appears to be independent of TLR, MEK and JAK signaling. Instead, we identified the T4SS and TNFα as being necessary for early activation of MAP kinase p38 and subsequent SOCS3 expression. Silencing studies revealed that SOCS3 expression limits H. pylori-induced secretion of pro- as well as anti-inflammatory cytokines by DCs while it specifically dampens PD-L1 expression but not co-stimulatory molecules. Accordingly, we demonstrate that SOCS expression in DCs is important for subsequent T-cell proliferation. Taken together, this study identifies SOCS3 as an important player in the induction ofH. pylori-induced immune responses.
Methods
Generation of monocyte-derived dendritic cells (DCs)
Monocyte-derived dendritic cells (moDCs) were generated from monocytes isolated from fresh buffy coats of healthy, anonymous donors (Blood Bank Salzburg, Austria) as described previously [23]. Briefly, peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Histopaque-1077 (Sigma). After erythrocyte lysis with ACK buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.4), monocytes were isolated by means of magnetic labeling using CD14 Microbeads Ultrapure (MACS Miltenyi) or by monocyte adherence (70 min at 37 °C, 5% CO2), and cultured in RPMI 1640 supplemented with 10% heat-inactivated fetal calf serum, 1% L-glutamine (PAA), 10 U/ml penicillin/streptomycin (Sigma), 50 μM β-mercaptoethanol (Gibco). Immature DCs were generated by supplementation of the medium with 50 ng/ml GM-CSF and IL-4, respectively, for 7 days. After 2 days of differentiation, fresh medium was added to the culture. After 7 days, DCs were harvested and seeded in 24-well plates for infection.
Bacterial culture and infection experiments
H. pylori wild-type P12 (wt) and P12 ΔcagPAI were cultured at 37 °C on GC agar plates (10% horse serum) under microaerophilic conditions (CampyGen Atmosphere Generation Systems, Thermo Scientific). For P12 ΔcagPAI, selective GC agar plates supplemented with kanamycin (8 μg/ml) were used. For infection, H. pylori was harvested and added to the DCs at a multiplicity of infection (MOI) of 0.2–20 or MOI = 5, respectively, for the indicated time points. For TLR inhibition experiments, DCs were treated with α-TLR2 (PAb-hTLR2, InvivoGen) and α-TLR4 (W7C11, α-hTLR4-IgG, InvivoGen) (1 μg/ml each) 20 min prior to infection. As a control, DCs were stimulated with 1 ng/ml LPS (E. coli). For the MAPK inhibition experiments, cells were treated with specific inhibitors of MEK1/2 (U0126, InvivoGen) or p38 (SB203, InvivoGen) at a concentration of 10 μM 1 h prior to infection. JAK kinase inhibition was performed by addition of a specific inhibitor (CAS 457081–03-7, Calbiochem, 1 μg/ml) 20 min prior to infection. TNFα was neutralized by using a human neutralizing antibody (D1B4, Cell Signaling) at a concentration of 100 ng/ml.
Flow cytometry and ELISA
DC phenotypes were determined by analysis of median fluorescence intensities of surface marker expression measured on a FACS Canto II flow cytometer (BD Biosciences). Cells were harvested and resuspended in FACS buffer (PBS, 1% BSA, 2 mM EDTA) before staining (30 min, 4 °C, dark) using the following antibodies: CD1a-BV421 (HI149), CD40-FITC (SC3) and CD86-PE (IT2.2) (eBioscience), CD80-APC-H7 (L307.4) and PD-L1-PE-Cy7 (MIH1) (BD Biosciences), HLA-DR-APC (LN3, Invitrogen), PD-L2-APC-Vio770 (Miltenyi) as well as LD (eBioscience Fixable Viability Dye eFluor 780 and eFluor 506). Analysis of the median fluorescence intensity of the mentioned markers within the living CD1a+ moDC population was performed with FlowJo Software. Cytokine ELISA in supernatants of infected cells was performed according to the manufacturer’s instructions: IL-1β (R&D) and IL-6, IL-8, IL-12, TNF-α (all Peprotech).
Multiplex assay
Cytokine and chemokine secretion of infected DCs was analyzed using the Cytokine/Chemokine/Growth Factor 45-Plex Human ProcartaPlex™ from ThermoFisher. Briefly, beads were washed once (PBS, 0.05% Tween-20) and resuspended in assay buffer (PBS, 0.05% Tween-20, 1% heat-inactivated FCS) before 8.34 μl per well were added into a 96-well V-bottom plate. Subsequently, 15 μl of standard or samples were added and the plate was incubated on an orbital shaker at 4 °C overnight. The next day, samples were washed three times and resuspended in 15 μl of detection antibody solution and incubated for 30 min at room temperature. Samples were again washed three times before 20 μl of Streptavidin-PE solution (1:1 in assay buffer) were added to each well and incubated for 30 min at room temperature. Eventually, samples were washed three times and resuspended in drive fluid for analysis. Measurement was performed on a Luminex Magpix instrument and data were analyzed using Procarta Plex Analyst Software (ThermoFisher).
RNA isolation and quantitative real-time PCR
Total RNA was isolated using TRI Reagent (Sigma) and reverse-transcribed with RevertAid H Minus M-MulV reverse transcriptase (Thermo Scientific) according to the manufacturer’s instructions. Expression levels were determined by quantitative real-time PCR on a Rotorgene 3000 (Corbett Research) using IQ SYBR Green Supermix. Levels of the large ribosomal protein P0 (RPLP0) served as a reference gene. Relative mRNA expression x was calculated using the equation x = 2-ΔCt, where Δct represents the difference between the threshold cycle (ct) of the gene of interest and the reference gene. PCR specificity was assessed by recording a melting curve for the PCR products. The following primer pairs were used for quantification:SOCS1: sense 5′-TTGGAGGGAGCGGATGGGTGTAG- 3′, antisense 5′-AGAGGTAGGAGGTGCGAGTTCAGGTC-3′; SOCS2: sense 5′-CCAAATCAACCAAAAAAAGTGACCATGAAGTCCTG-3′, antisense 5′- CGGGGATTGAGTTGCACCTGTATAGCATGATATTC-3′; SOCS3: sense 5′- ATACTATACCTTCCTGTACCTGGGTGGATGGAGCG-3′, antisense 5′- TGAGTATGTGGCTTTCCTATGCTGGGTCCCTCT-3′;
Database analysis
For analysis of SOCS expression in gastritis patients, the public genomic dataset GSE5081 from NCBI Gene Expression Omnibus (NCBI-GEO) [29] was used. The data discussed in the present study were derived from Galamb et al. [30], have been deposited in NCBI-GEO, and are accessible through GEO Series accession number GSE5081. Analysis was performed using Python. Access to the GEO database was granted with the following GEOparse package (https://github.com/guma44/GEOparse). The GSE5081 dataset includes whole-genome oligonucleotide microarray analysis data of H. pylori-related (HP+) and idiopathic (HP-) gastritis patients. Gene expression data derive from total RNA which was extracted from frozen gastric biopsy specimens. The dataset includes 32 samples of 16 patients (8 HP+, 8 HP-), in which erosive and non-erosive (normal antrum) areas were used for biopsies. While SOCS1 and SOCS2 gene expression data show no statistical difference, SOCS3 expression does show a statistically significant difference between HP+ and HP- samples (Affymetrix Probe Set IDs in Platform GPL570: 213337_s_at, 232539_at and 227697_at).
Western blot
Cell pellets of infected samples were harvested and lysed in 80 μl 2x Laemmli sample buffer (BIO-RAD) supplemented with 5% β-mercaptoethanol (Sigma). Samples were separated on a 4–12% gradient gel (NuPAGE, Life Technologies) and blotted onto a nitrocellulose membrane (BIO-RAD). Membranes were blocked in TBS supplemented with 0.1% Tween and 5% nonfat dry milk for 1 h. The following antibodies were used according to the manufacturer’s instructions: p-STAT1-Tyr701 (Cat no. 5375S), STAT-1 (Cat no. 9172S), p-STAT3-Tyr705 (Cat no. 9145S), STAT3 (Cat no. 4904S), p-p38-Thr180/Tyr182 MAPK (Cat no. 9215S), p38 (Cat no. 9212S), and HRP-linked anti-rabbit secondary antibody (Cat no. 7074) (all Cell Signaling Technology). Detection was done using West Pico PLUS Chemiluminescent Substrate (Thermo Fisher) and BioMax films (Kodak). Blots were quantified using ImageJ (NIH) software [31].
Gene silencing via siRNA-based transfection
DCs were transfected with small interfering RNAs (siRNAs) targetingSOCS1 (Santa Cruz Biotechnology, Inc.),SOCS2 (ThermoFisher), SOCS3 (ThermoFisher) or Allstars negative control (Qiagen). For transfection, Lipofectamine RNAiMAX reagent (Life Technologies) was used according to the manufacturer’s instructions. In brief, 2 × 105 DCs were seeded in 100 μl DC medium and transfected with 100 μl Opti-MEM (Life Technologies) containing 50 to 100 pmol siRNA and 1 μl transfection reagent. Subsequently, DCs were incubated for 6 h before 800 μl of fresh DC medium was added. After 48 h of incubation, DCs were used for infection experiments after silencing efficiency was assessed by qRT-PCR.
Allogeneic moDC/T-cell co-culture
Control or SOCS3-silenced moDCs were harvested after 48 h and replated at a density of 105 cells/400 μl in P/S free T-cell medium (IMDM + 5% FCS, 1x L-Glut). Thereafter, moDCs were either infected with H. pylori or left untreated for 24 h. The next day, 6 h pre addition of T cells, the cultures were supplemented with P/S (1x). Total CD4+ T cells were isolated from buffy coats of healthy donors using the Human CD4+ T cell isolation kit (Miltenyi Biotec) according to the manufacturer’s instructions, and isolated T cells were stained with e450 Proliferation Dye (eBioscience) for 10 min at room temperature. Thereafter, T cells were added to the moDC culture at a ratio of 1:10 (moDC:T cell). IL-2 was added at a concentration of 50 U/ml. After 6 days of co-culture, T cells were stimulated with PMA (50 ng/ml), Ionomycin (1 μg/ml) and Brefeldin A (10 μg/ml, Sigma-Aldrich B652) for 4 h. Thereafter, cells were transferred in a V-bottom plate and stained in 20 μl PBS containing the following antibodies for surface staining: CD4-PE-594 (RPT-4A, BioLegend) and LD (eBioscience Fixable Viability Dye eFluor 780) for 30 min (4 °C, dark). After washing of the samples (150 μl PBS), 100 μl Fix-Perm (FOXP3 staining kit, eBioscience) were added and the samples were incubated for 30 min (4 °C, dark). Meanwhile, the intracellular staining mix was prepared in PermBuffer using IFNγ-PE-Cy7 (B27). After two washing steps, IFNγ-PE-Cy7 (B27) diluted in PermBuffer was added to the samples for 20 min. Measurement was performed in PBS + 2 mM EDTA on a CytoFLEX S instrument (Beckman Coulter). Living, single, CD4+ T cells were used to analyze IFNγ levels.
Statistics
Data are presented as bars indicating mean ± standard deviation (SD) or as dot plots. Statistical analyses were performed with GraphPad Prism 7 software. Multiple groups were analyzed by one-way ANOVA including a post-hoc test. P values < 0.05 were considered significant (*p < 0.05, **p < 0.01, ***p < 0.001, n.s. not significant).
Results
H. pylori induces cytokine secretion and expression of feedback inhibitors in a time-dependent way
H. pylori has been shown to penetrate the gastric lining and directly interact with cells of the immune system. Immature DCs are recruited to the inflamed mucosa where they encounter the bacteria and undergo maturation, which initiates an adaptive immune response toward the pathogen [32]. In general, the DC maturation process involves the release of inflammatory cytokines and elevated levels of co-stimulatory molecules, which are crucial for T-cell activation [33, 34]. Accordingly, we observed dose-dependent upregulation of activation markers as well as cytokine secretion upon H. pylori infection (Supplementary Figure 1). It is well established that bacterial stimuli also induce the expression of immunosuppressive molecules, including programmed death ligand 1 (PD-L1) and PD-L2, in order to prevent excessive damage to the tissue following an inflammatory response [35, 36]. Consistent with this, we observed significant and dose-dependent increases of both PD-L1 and PD-L2 levels upon H. pylori infection (Supplementary Figure 1A).
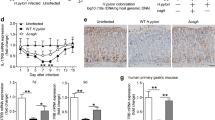
To examine the inflammatory response of DCs to H. pylori infection in more detail, we monitored cytokine and chemokine secretion upon infection over a defined time period (Fig. 1a). We observed the release of a wide variety of mediators, with IL-6 and TNFα being significantly increased at 8 h post-infection with H. pylori, whereas most other cytokines were detected only after 24 h (Fig. 1a/b). In light of these strong increases in cytokine levels, we additionally investigated the mRNA expression of suppressor of cytokine signaling (SOCS) proteins SOCS1, − 2 and − 3 over a period of 48 h post H. pylori infection (Fig. 1c). Our data reveal that SOCS3 mRNA was already induced within 1 h of infection, whereasSOCS1 and SOCS2 expression was delayed and detectable only after 8 h. Neither H. pylori infection nor TLR4 stimulation with E. coli LPS was able to induce SOCS2 expression within 4 h of treatment (Fig. 2a), which is in line with previous studies demonstrating the delayed expression profile of SOCS2 [23]. Interestingly, LPS significantly induced the expression of both SOCS1 and SOCS3 at this early time point, whereas H. pylori selectively increased only SOCS3 expression (Fig. 2a). Additionally, we analyzed SOCS expression in a data set ofH. pylori-positive and negative human gastric biopsies and found that SOCS3 was significantly increased in H. pylori-positive samples compared to H. pylori-negative ones, while SOCS1 and SOCS2 were not differentially expressed (Fig. 2b). The exclusive and rapid induction ofSOCS3 suggests that SOCS3 might play a specific role in regulating DC activation in response to H. pylori infection.

H. pylori infection elicits cytokine and chemokine secretion as well as SOCS expression in moDCs. DCs were re-plated in fresh medium,H. pylori strain P12 was harvested in PBS and added to the cells at MOI = 5. a Heatmap depicting cytokine and chemokine secretion by DCs infected for the indicated times, ranging from 1 to 48 h, analyzed by Multiplex Assay. b Detailed presentation of secretion of IL-6, IL-8, IL-12 and TNFα by infected moDCs in a time-dependent manner. Each dot represents an individual donor; the horizontal line in each column indicates the mean value of one experiment comprising 4 donors. (N = 4) cSOCS1, SOCS2, SOCS3 mRNA expression was monitored by qRT-PCR. Bars represent mean + SD of three experiments comprising six individual donors. (N = 6) For statistical analysis, one-way ANOVA with a Sidak post-hoc test was performed. *p < 0.05, **p < 0.01, ***p < 0.001

H. pylori selectively augments SOCS3 expression.a DCs were infected withH. pylori strain P12 at MOI = 5 or stimulated with E. coli-derived LPS (1 ng/ml) for 4 h. SOCS mRNA expression was measured by qRT-PCR. Dots represent individual donors; the horizontal line in each column indicates the mean value ± SD of one experiment comprising four donors. (N = 4) For statistical analysis, one-way ANOVA with Tukey’s post-hoc test was performed. **p < 0.01, ***p < 0.001 b SOCS expression in H. pylori-negative and H. pylori-positive gastritis patients was determined using a publicly available genomic dataset (GSE5081), which was analyzed using Python. Dots represent individual donors; the horizontal line in each column indicates the mean value ± SD. (N = 16) For statistical analysis, a two-tailed, unpaired t test was performed. *p < 0.05
H. pylori triggers early SOCS3 expression via p38 signaling but independent of TLR, MEK1/2 and JAK
To determine how H. pylori induces SOCS3 expression within 1 h of infection, we first investigated whether activation of TLRs might contribute to this gene activation. In particular, TLR2 and TLR4 have been reported to be involved in sensing H. pylori products, which in turn leads to DC activation [19]. Therefore, we treated DCs with a combination of α-TLR2 and α-TLR4 blocking antibodies prior to infection and monitored subsequent expression of SOCS proteins. To control for successful TLR blocking, DCs were treated with E. coli LPS, a well-described ligand of TLR2 and TLR4 [37, 38]. Accordingly, we confirmed that addition of TLR2/4 blocking antibodies potently blocked E. coli LPS-induced mRNA expression of SOCS3 by DCs (Fig. 3a, right panel). In contrast, H. pylori-induced SOCS3 expression was not affected by the addition of the TLR blocking antibodies (Fig. 3a, left panel). These data demonstrate that, within the first hour of infection, H. pylori-induced SOCS3 expression was independent of TLR2/4 signaling. Because JAK activation and subsequent STAT signaling are well-described mechanisms driving SOCS expression, we next determined whether this pathway may contribute to early SOCS3 regulation [39]. Similar to our previous inhibition experiments, JAK inhibition did not have an impact on H. pylori-induced SOCS3 expression (Fig. 3b). Therefore, we focused on mitogen-activated protein (MAP) kinases, which have been shown to be rapidly activated by H. pylori in epithelial cells [40] and to play a role in regulation of SOCS3 mRNA levels [41]. To test whether these kinases are potential inducers of SOCS3 in DCs, we blocked p38 and MEK1/2 prior to infection and monitored SOCS3 expression 1 h post infection. While inhibition of MEK1/2 had no effect on SOCS3 induction (Supplementary Figure 2), blocking of p38 decreased the capacity of H. pylori to induce SOCS3 (Fig. 3c). These data suggest that MAPK p38 is crucially involved in H. pylori-induced SOCS3 expression within the first hour of infection.

H. pylori-inducedSOCS3 expression is mediated by p38 and independent of TLR2/4 and JAK kinase. After 1 h of infection with H. pylori at MOI = 5 or stimulation with LPS, DCs were harvested and SOCS3 mRNA expression was analyzed by qRT-PCR. a 20 min prior to infection with H. pylori or stimulation with LPS (1 ng/mL), blocking antibodies for TLR2 (1 μg/mL) and TLR4 (1 μg/mL) were added to the cells (αTLR2/4). H. pylori strain P12 was added to the cells at MOI = 5 (orange). LPS was added at 1 ng/mL (blue). One experiment comprising 3 individual donors is shown. (N = 3) b, c Specific inhibitors of JAK kinase (1 μg/ml) or p38 (SB203) (10 μM) were added to the respective samples. Dots represent individual donors; mean ± SD is shown. b Four experiments comprising 9 donors (N = 9) and c three experiments comprising 6 donors (N = 6) are shown. For statistical analysis, one-way ANOVA with Tukey’s post-hoc test was performed. *p < 0.05, **p < 0.01
H. pylori wild type but not the ΔCagPAI mutant induces SOCS3 expression via p38
To confirm the role of MAPK p38 in H. pylori-induced SOCS3 expression, we infected DCs and monitored the phosphorylation of p38 (p-p38) during the first 2 h of H. pylori infection. We detected phosphorylated p38 as early as 30 min post-infection, whereas phosphorylation of the canonical SOCS3-inducer STAT3 [42] as well as STAT1 was observed only after 2 h (Fig. 4a). This further supports our hypothesis that the p38 MAPK pathway is involved in H. pylori-induced SOCS3 expression, whereas STAT3 plays a minor role in this context. Because we already excluded TLR2 and TLR4 as triggers of early SOCS3 expression (Fig. 3a), we next addressed the question of whether the CagPAI-encoded type IV secretion system (T4SS) is necessary for p38 phosphorylation and thus for early induction of SOCS3. We infected DCs of three independent donors with either the H. pylori wild-type (wt) strain P12 or a P12 mutant lacking CagPAI (ΔCagPAI) and analyzed p38 phosphorylation. While the H. pylori wt strain strongly induced phosphorylation of p38, the T4SS-deficient strain ΔCagPAI showed severely impaired p38 activation (Fig. 4b). We additionally analyzedSOCS3 expression in cells infected withH. pylori wt or the ΔCagPAI mutant and observed significantly lower SOCS3 mRNA levels upon infection with the mutant compared with the wt strain (Fig. 4c). These results indicate that both the T4SS and p38 are necessary for H. pylori-induced early SOCS3 expression.

H. pylori triggers early phosphorylation of p38 which correlates with SOCS3 expression. Day-7 DCs were re-plated in fresh medium, H. pylori strain P12 was harvested in PBS and added to the cells at MOI = 5. a Protein levels and phosphorylation of p38, STAT1 and STAT3 were analyzed at the indicated time points by western blotting. One representative experiment out of two is shown. b DCs were infected with H. pylori strain P12 wt or ΔCagPAI, and p-p38 levels were detected after 30 min. Three individual donors are shown. c DCs were infected with H. pylori strain P12 wt or ΔCagPAI, and SOCS3 mRNA expression was analyzed after 1 h. Dots represent individual donors, mean ± SD of eight individual donors is shown. For statistical analysis, one-way ANOVA with Tukey’s post-hoc test was performed. ***p < 0.001
Early SOCS3 expression is a consequence of T4SS-dependent release of TNFα
As a next step we investigated whether H. pylori infection per se or mediators released upon H. pylori infection drive SOCS3 expression. Interestingly, we observed that H. pylori wt-induced SOCS3 expression was significantly inhibited when cytokine release was blocked using Brefeldin A (Fig. 5a). Treatment with Brefeldin A results in SOCS3 expression comparable to that observed upon infection with the ΔCagPAI mutant. This suggests that infection with the ΔCagPAI mutant does not induce the secretion of the soluble mediator crucial forSOCS3 expression (Fig. 5a). As IL-6 and TNFα are both known to enhanceSOCS3 expression, we monitored gene expression of these cytokines in the same experimental setting. Intriguingly, the ΔCagPAI mutant was able to induce expression of IL-6 but failed to induce expression of TNFα after 1 h of infection, whereas Brefeldin A treatment resulted in decreased IL-6 but stable TNFα mRNA levels upon infection with the wt strain (Fig. 5b). These results suggest a potential role for TNFα in mediating H. pylori-induced SOCS3 mRNA expression. To confirm this hypothesis, we induced moDCs with TNFα to analyze its capacity to induce SOCS3 mRNA expression and, additionally, we blocked TNFα in DCs using a neutralizing antibody prior to infection. We found that TNFα stimulation results in SOCS3 mRNA expression, which could be blocked upon neutralization of TNFα (Fig. 5c). Interestingly, blocking of TNFα also suppressed H. pylori wt-induced SOCS3 expression (Fig. 5c). Taken together, this set of data suggests that only H. pylori wt, but not the ΔCagPAI mutant, is able to induce TNFα expression within 1 h of infection, which in turn activates the expression of SOCS3 mRNA.

SOCS3 expression is a secondary response to H. pylori-induced TNFα. After 1 h of infection withH. pylori at MOI = 5, DCs were harvested and mRNA expression was analyzed by qRT-PCR.a, b 1 h prior to infection with H. pylori strain P12 WT (orange) or ΔCagPAI (green), Brefeldin A (1 μg/ml) was added to the cells. H. pylori was added to the cells at MOI = 5 and SOCS3, IL6 and TNFα mRNA expression was evaluated after 1 h. Two experiments comprising 6 individual donors (N = 6) are shown. Dots represent individual donors; mean ± SD is shown. c Cells were treated with a specific TNFα neutralizing antibody (100 ng/ml) before infection with H. pylori strain P12 WT or stimulation with TNFα (10 ng/ml). After 1 h, SOCS3 mRNA expression was assessed. One experiment comprising 5 individual donors (N = 5) is shown. Dots represent individual donors; mean ± SD is shown. For statistical analysis, one-way ANOVA with Tukey’s post-hoc test was performed. *p < 0.05, **p < 0.01
Silencing of SOCS3 in DCs results in enhanced PD-L1 expression and dampened CD4+ T-cell proliferation
Having shown that H. pylori induces the expression of SOCS3 within the first hour post infection, we analyzed whether the presence of SOCS3 affects H. pylori-induced activation of human DCs. To investigate the role of SOCS3 during H. pylori infection, we performed RNA interference-based silencing of SOCS3 expression prior to infection. Transfection with siRNA targeting SOCS3 effectively reduced SOCS3 expression in DCs infected with H. pylori, compared to DCs treated with a non-targeting control siRNA (Fig. 6a). We then measured H. pylori-induced secretion of soluble mediators by DCs. The secretion of several pro-inflammatory and anti-inflammatory cytokines, including IL-1β, IL-12 and TNFα, IL-27 and IL-1RA, was augmented upon H. pylori infection of DCs transfected with siRNA targeting SOCS3 (Fig. 6b/c). Interestingly, SOCS3 silencing also resulted in a significant increase of PD-L1, whereas CD40, CD80, CD86 and PD-L2 remained unchanged, compared to control cells (Fig. 6d and Supplementary Figure 3). In contrast, silencing ofSOCS1 and SOCS2, respectively, did not significantly alter the DC phenotype upon infection (Supplementary Figure 4). These data indicate that H. pylori-induced SOCS3 regulates inflammatory cytokine release, but it also dampens the secretion of anti-inflammatory mediators and the induction of the co-inhibitory molecule PD-L1, which could result in persistent inflammation. Therefore, the question arose whether SOCS3 expression in DCs also boosts H. pylori-induced T-cell responses. To address this question, we performed allogeneic co-cultures with SOCS3-silenced moDCs and CD4+ T cells and analyzed proliferation as well as IFNγ expression. We observed significantly decreased T-cell proliferation and IFNγ expression by CD4+ T cells upon co-culture withSOCS3-silenced H. pylori-infected cells compared to control silenced cells. Therefore, these data indicate that H. pylori induced-SOCS3 expression in DCs limits PD-L1 levels, which in turn results in increased T-cell proliferation.

SOCS3 silencing shapes H. pylori-induced DC activation and promotes proliferation of T cells. Immature day-7 DCs were re-plated and transfected with siRNA targetingSOCS3 or non-targeting control oligo (both 100 pmol). 48 h post-transfection, H. pylori strain P12 was harvested in PBS and added to the cells at MOI = 5. a Silencing efficiency was analyzed by qRT-PCR after 2 h of infection. One experiment comprising four individual donors is shown. (N = 4) b/c Cytokine secretion of three experiments comprising eight donors (N = 8) was analyzed 24 h post-infection. d Surface marker expression of two experiments comprising six donors (N = 6) was analyzed 24 h after bacterial infection. e/f Control (si Ctrl) or SOCS3-silenced (si SOCS3) DCs were infected with H. pylori (MOI = 5) for 24 h before P/S was added for 6 h. Total CD4+ T cells were magnetically isolated from PBMCs and cultivated with DCs at a ratio of 1:10 (DC:T cell) for 6 days. On day 6, cells were restimulated with PMA/Ionomycin for 4 h and analyzed by means of flow cytometry. Dots represent individual donors (N = 3); mean ± SD is shown. For statistical analysis, one-way ANOVA with Tukey’s post-hoc test was performed. *p < 0.05, **p < 0.01
Discussion
Numerous studies have shown that H. pylori and soluble factors associated with this pathogen are potent activators of innate immune cells, as characterized by the release of pro-inflammatory mediators and expression of co-stimulatory molecules [43,44,45]. However, the immune response towardH. pylori fails to effectively clear the pathogen, leading to lifelong infection within the human stomach. Manifold evolved strategies to escape and manipulate the host’s innate and adaptive immunity makeH. pylori a very successful pathogen. These mechanisms range from circumventing phagocytic activity and recognition by innate immune cells [17, 46] to inducing adaptive immune responses with a regulatory profile [47,48,49].
Although it is well established that immune cells drive the expression of negative feedback regulators (e.g. SOCS proteins) to limit excessive immune responses upon bacterial encounters [50, 51], evidence for a role of SOCS proteins during H. pylori infection remains scarce. As we are interested in the early events in DC activation critical for priming the signaling network of these cells, we investigated the transcriptional activation of negative feedback inhibitors at early time points after H. pylori infection. Our data indicate a specific and strong upregulation of SOCS3 after 1 h of infection, while SOCS1 and SOCS2 displayed delayed kinetics. To identify the pathway involved in SOCS3 transcription, we analyzed TLR signaling as well as the activation of MAPKs and STAT proteins, as both p38 and STAT3 have been linked to transcriptional regulation of SOCS3 [42, 52, 53]. We demonstrated that early induction of SOCS3 by H. pylori is dependent on an intact T4SS as well as MAPK p38, but independent of TLR2, TLR4, MEK1/2 and the JAK/STAT pathway. This raises the question whether p38 phosphorylation and SOCS3 expression are stimulated by a specific cytokine milieu created immediately upon infection withH. pylori wt. Accordingly, we could demonstrate that H. pylori wt, but not the ΔCagPAI mutant, directly activates transcription of TNFα, which in turn results in upregulation of SOCS3. These data are in line with a study describing that p38 activation via TNFα enhances SOCS3 mRNA stabilization and thus its expression in murine fibroblasts [41]. However, it remains unclear whether TNFα expression, and thus p38-mediated SOCS3 expression, is stimulated by structural components of the T4SS or by injected factors. In this regard, quantitative phospho-proteomics data suggest that phosphorylation of p38 is independent of cagA translocation [54]. Accordingly, newer studies show that the LPS heptose core molecule ADP heptose is also injected via the T4SS into infected cells and triggers TIFA-dependent p38 signaling [55, 56].
As mentioned above, H. pylori specifically increases SOCS3 at early time points. This is in contrast to stimulation of DCs with conventional E. coli LPS, where we observed rapid upregulation of both SOCS1 and SOCS3. The synchronized regulation of SOCS1 and SOCS3 expression in DCs and macrophages has been reported by numerous studies employing purified TLR agonists, like LPS or CpG [57, 58], but also in response to bacterial infection with pathogens, including Salmonella enterica, Chlamydia pneumoniae and Mycobacterium tuberculosis [59,60,61]. In this respect, SOCS1 expression in macrophages has been implicated in hampering of bacterial clearance during Mycobacterium tuberculosis and Chlamydia pneumoniae infection while also protecting the host from excessive pathogen-induced inflammation [61]. However, the role of SOCS3 in innate inflammation is less well understood and our observation that H. pylori specifically induces SOCS3 expression raises the question of the consequences of selective SOCS3 expression. Murine bone-marrow-derived DCs overexpressing SOCS3 were shown to express low levels of MHC II and CD86, and low levels of IL-12 and IL-23, upon stimulation with LPS [28]. Consistent with this, we showed that human DCs lackingSOCS3 secrete higher amounts of IL-12, IL-23 and IL-1β, while we also detected increased expression of the anti-inflammatory cytokine IL-1RA. We also observed increased H. pylori-induced secretion of TNFα and IL-27 as well as enhanced levels of PD-L1 upon SOCS3 silencing. The upregulation of PD-L1 might be a result of pronounced TNFα and/or IL-27 secretion, as recent studies described that both cytokines support PD-L1 expression [62, 63]. Whereas TNFα was shown to enhance PD-L1, but not PD-L2, in a NF-κB-dependent manner [62], IL-27 stimulation was found to be related to PD-L1 upregulation in various immune cells, including human monocytes and DCs [63]. Additionally, SOCS3 overexpression has been reported to result in decreased levels of IL-27-induced PD-L1 expression [64], which suggests that SOCS3 plays a role in limiting PD-L1 expression. To test whether DC-specific SOCS3 deficiency also influences consequential T-cell responses, we performed co-culture experiments where we observed decreased T-cell proliferation as well as decreased IFNγ secretion by CD4+ T cells upon SOCS3 silencing. This observation is in line with a study from Gao et al., who demonstrated that in a model of Mycobacterium tuberculosis infection, antigen-presenting cells lacking SOCS3 were poor inducers of mycobacterial-specific T-cell responses [65].
Conclusions
In conclusion, we demonstrated that H. pylori regulates SOCS3 during early infection and that this process requires an intact T4SS system and TNFα-mediated activation of p38. Our results further suggest that hijacking of DC signaling cascades and consequential upregulation of SOCS3 modulates T-cell responses upon H. pylori infection.
Availability of data and materials
The datasets used and/or analyzed in the present study are available from the corresponding author on reasonable request.
Abbreviations
- DC:
-
Dendritic cell
- MAPK:
-
Mitogen-activated protein kinase
- moDC:
-
Monocyte-derived dendritic cell
- MOI:
-
Multiplicity of infection
- PBMC:
-
Peripheral blood mononuclear cell
- PD-L1/2:
-
Programmed death ligand 1/2
- SOCS:
-
Suppressor of cytokine signaling
- STAT:
-
Signal transducer and activator of transcription
- TLR:
-
Toll-like receptor
- T4SS:
-
Type IV secretion system
References
Schreiber S, Konradt M, Groll C, Scheid P, Hanauer G, Werling H-O, Josenhans C, Suerbaum S. The spatial orientation of <em>helicobacter pylori</em> in the gastric mucus. Proc Natl Acad Sci U S A. 2004;101:5024–9.
Morris AJ, Ali MR, Nicholson GI, Perez-Perez GI, Blaser MJ. Long-term follow-up of voluntary ingestion of helicobacter pylori. Ann Intern Med. 1991;114:662–3.
Graham DY, Opekun AR, Osato MS, El-Zimaity HM, Lee CK, Yamaoka Y, Qureshi WA, Cadoz M, Monath TP. Challenge model for helicobacter pylori infection in human volunteers. Gut. 2004;53:1235–43.
Kuipers EJ, Israel DA, Kusters JG, Gerrits MM, Weel J, van Der Ende A, van Der Hulst RW, Wirth HP, Hook-Nikanne J, Thompson SA, Blaser MJ. Quasispecies development of helicobacter pylori observed in paired isolates obtained years apart from the same host. J Infect Dis. 2000;181:273–82.
Peek RM Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37.
Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784–9.
Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of helicobacter pylori infection. Clin Microbiol Rev. 2006;19:449–90.
Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7–14 June 1994. IARC Monogr Eval Carcinog Risks Hum 1994, 61:1–241.
Posselt G, Backert S, Wessler S. The functional interplay of helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun Signal. 2013;11:77.
Pfannkuch L, Hurwitz R, Traulsen J, Sigulla J, Poeschke M, Matzner L, Kosma P, Schmid M, Meyer TF. ADP heptose, a novel pathogen-associated molecular pattern identified in helicobacter pylori. FASEB J. 2019;33:9087–99.
Xu XQ, Wang ZH, Liao JX, Chen XY, Liu WZ, Xiao SD, Lu H. Predictive value of neutrophil infiltration as a marker of helicobacter pylori infection. World J Gastroenterol. 2012;18:5101–5.
Nurgalieva ZZ, Conner ME, Opekun AR, Zheng CQ, Elliott SN, Ernst PB, Osato M, Estes MK, Graham DY. B-cell and T-cell immune responses to experimental helicobacter pylori infection in humans. Infect Immun. 2005;73:2999–3006.
Bimczok D, Clements RH, Waites KB, Novak L, Eckhoff DE, Mannon PJ, Smith PD, Smythies LE. Human primary gastric dendritic cells induce a Th1 response to H. pylori. Mucosal Immunol. 2010;3:260–9.
Khamri W, Walker MM, Clark P, Atherton JC, Thursz MR, Bamford KB, Lechler RI, Lombardi G. Helicobacter pylori stimulates dendritic cells to induce interleukin-17 expression from CD4+ T lymphocytes. Infect Immun. 2010;78:845–53.
Necchi V, Manca R, Ricci V, Solcia E. Evidence for transepithelial dendritic cells in human H. pylori active gastritis. Helicobacter. 2009;14:208–22.
Shiu J, Blanchard TG. Dendritic cell function in the host response to helicobacter pylori infection of the gastric mucosa. Pathog Dis. 2013;67:46–53.
Andersen-Nissen E, Smith KD, Strobe KL, Barrett SL, Cookson BT, Logan SM, Aderem A. Evasion of toll-like receptor 5 by flagellated bacteria. Proc Natl Acad Sci U S A. 2005;102:9247–52.
Cullen TW, Giles DK, Wolf LN, Ecobichon C, Boneca IG, Trent MS. Helicobacter pylori versus the host: remodeling of the bacterial outer membrane is required for survival in the gastric mucosa. PLoS Pathog. 2011;7:e1002454.
Kabisch R, Mejias-Luque R, Gerhard M, Prinz C. Involvement of toll-like receptors on helicobacter pylori-induced immunity. PLoS One. 2014;9:e104804.
Duncan SA, Baganizi DR, Sahu R, Singh SR, Dennis VA. SOCS proteins as regulators of inflammatory responses induced by bacterial infections: a review. Front Microbiol. 2017;8:2431.
Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–65.
Bartz H, Avalos NM, Baetz A, Heeg K, Dalpke AH. Involvement of suppressors of cytokine signaling in toll-like receptor-mediated block of dendritic cell differentiation. Blood. 2006;108:4102–8.
Posselt G, Schwarz H, Duschl A, Horejs-Hoeck J. Suppressor of cytokine signaling 2 is a feedback inhibitor of TLR-induced activation in human monocyte-derived dendritic cells. J Immunol. 2011;187:2875–84.
Neuper T, Ellwanger K, Schwarz H, Kufer TA, Duschl A, Horejs-Hoeck J. NOD1 modulates IL-10 signalling in human dendritic cells. Sci Rep. 2017;7:1005.
Shen L, Evel-Kabler K, Strube R, Chen SY. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat Biotechnol. 2004;22:1546–53.
Shi D, Li D, Yin Q, Qiu Y, Yan H, Shen Y, Lu G, Liu W. Silenced suppressor of cytokine signaling 1 (SOCS1) enhances the maturation and antifungal immunity of dendritic cells in response to Candida albicans in vitro. Immunol Res. 2015;61:206–18.
Liu X, Qu X, Chen Y, Liao L, Cheng K, Shao C, Zenke M, Keating A, Zhao RC. Mesenchymal stem/stromal cells induce the generation of novel IL-10-dependent regulatory dendritic cells by SOCS3 activation. J Immunol. 2012;189:1182–92.
Li Y, Chu N, Rostami A, Zhang GX. Dendritic cells transduced with SOCS-3 exhibit a tolerogenic/DC2 phenotype that directs type 2 Th cell differentiation in vitro and in vivo. J Immunol. 2006;177:1679–88.
Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–10.
Galamb O, Gyorffy B, Sipos F, Dinya E, Krenacs T, Berczi L, Szoke D, Spisak S, Solymosi N, Nemeth AM, et al. Helicobacter pylori and antrum erosion-specific gene expression patterns: the discriminative role of CXCL13 and VCAM1 transcripts. Helicobacter. 2008;13:112–26.
Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5.
Kao JY, Rathinavelu S, Eaton KA, Bai L, Zavros Y, Takami M, Pierzchala A, Merchant JL. Helicobacter pylori-secreted factors inhibit dendritic cell IL-12 secretion: a mechanism of ineffective host defense. Am J Physiol Gastrointest Liver Physiol. 2006;291:G73–81.
Kranzer K, Eckhardt A, Aigner M, Knoll G, Deml L, Speth C, Lehn N, Rehli M, Schneider-Brachert W. Induction of maturation and cytokine release of human dendritic cells by helicobacter pylori. Infect Immun. 2004;72:4416–23.
Fehlings M, Drobbe L, Moos V, Renner Viveros P, Hagen J, Beigier-Bompadre M, Pang E, Belogolova E, Churin Y, Schneider T, et al. Comparative analysis of the interaction of helicobacter pylori with human dendritic cells, macrophages, and monocytes. Infect Immun. 2012;80:2724–34.
Brown JA, Dorfman DM, Ma F-R, Sullivan EL, Munoz O, Wood CR, Greenfield EA, Freeman GJ. Blockade of programmed Death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003;170:1257.
Wu YY, Lin CW, Cheng KS, Lin C, Wang YM, Lin IT, Chou YH, Hsu PN. Increased programmed death-ligand-1 expression in human gastric epithelial cells in helicobacter pylori infection. Clin Exp Immunol. 2010;161:551–9.
Dziarski R, Wang Q, Miyake K, Kirschning CJ, Gupta D. MD-2 enables toll-like receptor 2 (TLR2)-mediated responses to lipopolysaccharide and enhances TLR2-mediated responses to gram-positive and gram-negative bacteria and their cell wall components. J Immunol. 2001;166:1938–44.
Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem. 1999;274:10689–92.
Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117:1281.
Keates S, Keates AC, Warny M, Peek RM Jr, Murray PG, Kelly CP. Differential activation of mitogen-activated protein kinases in AGS gastric epithelial cells by cag+ and cag- helicobacter pylori. J Immunol. 1999;163:5552–9.
Ehlting C, Lai WS, Schaper F, Brenndorfer ED, Matthes RJ, Heinrich PC, Ludwig S, Blackshear PJ, Gaestel M, Haussinger D, Bode JG. Regulation of suppressor of cytokine signaling 3 (SOCS3) mRNA stability by TNF-alpha involves activation of the MKK6/p38MAPK/MK2 cascade. J Immunol. 2007;178:2813–26.
Zhang L, Badgwell DB, Bevers JJ 3rd, Schlessinger K, Murray PJ, Levy DE, Watowich SS. IL-6 signaling via the STAT3/SOCS3 pathway: functional analysis of the conserved STAT3 N-domain. Mol Cell Biochem. 2006;288:179–89.
Kranzer K, Sollner L, Aigner M, Lehn N, Deml L, Rehli M, Schneider-Brachert W. Impact of helicobacter pylori virulence factors and compounds on activation and maturation of human dendritic cells. Infect Immun. 2005;73:4180–9.
de Jonge R, Kusters J, Timmer SM, Gimmel V, Appelmelk B, Bereswill S, van Vliet A, GMS M, Kist M, Vandenbroucke-Grauls C, Kuipers E. The role of Helicobacter pylori virulence factors in interleukin production by monocytic cells; 2001.
Hafsi N, Voland P, Schwendy S, Rad R, Reindl W, Gerhard M, Prinz C. Human Dendritic Cells Respond to <em>Helicobacter pylori</em>, Promoting NK Cell and Th1-Effector Responses In Vitro. J Immunol. 2004;173:1249.
Wang G, Ge Z, Rasko DA, Taylor DE. Lewis antigens in helicobacter pylori: biosynthesis and phase variation. Mol Microbiol. 2000;36:1187–96.
Altobelli A, Bauer M, Velez K, Cover TL, Muller A. Helicobacter pylori VacA targets myeloid cells in the gastric lamina Propria to promote peripherally induced regulatory T-cell differentiation and persistent infection. MBio. 2019;10.
Oertli M, Noben M, Engler DB, Semper RP, Reuter S, Maxeiner J, Gerhard M, Taube C, Muller A. Helicobacter pylori gamma-glutamyl transpeptidase and vacuolating cytotoxin promote gastric persistence and immune tolerance. Proc Natl Acad Sci U S A. 2013;110:3047–52.
Wustner S, Mejias-Luque R, Koch MF, Rath E, Vieth M, Sieber SA, Haller D, Gerhard M. Helicobacter pylori gamma-glutamyltranspeptidase impairs T-lymphocyte function by compromising metabolic adaption through inhibition of cMyc and IRF4 expression. Cell Microbiol. 2015;17:51–61.
Latvala S, Miettinen M, Kekkonen RA, Korpela R, Julkunen I. Lactobacillus rhamnosus GG and Streptococcus thermophilus induce suppressor of cytokine signalling 3 (SOCS3) gene expression directly and indirectly via interleukin-10 in human primary macrophages. Clin Exp Immunol. 2011;165:94–103.
Baetz A, Frey M, Heeg K, Dalpke AH. Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J Biol Chem. 2004;279:54708–15.
Canfield S, Lee Y, Schroder A, Rothman P. Cutting edge: IL-4 induces suppressor of cytokine signaling-3 expression in B cells by a mechanism dependent on activation of p38 MAPK. J Immunol. 2005;174:2494–8.
Qin H, Roberts KL, Niyongere SA, Cong Y, Elson CO, Benveniste EN. Molecular mechanism of lipopolysaccharide-induced SOCS-3 gene expression in macrophages and microglia. J Immunol. 2007;179:5966–76.
Glowinski F, Holland C, Thiede B, Jungblut PR, Meyer TF. Analysis of T4SS-induced signaling by H. pylori using quantitative phosphoproteomics. Front Microbiol. 2014;5:356–6.
Stein SC, Faber E, Bats SH, Murillo T, Speidel Y, Coombs N, Josenhans C. Helicobacter pylori modulates host cell responses by CagT4SS-dependent translocation of an intermediate metabolite of LPS inner core heptose biosynthesis. PLoS Pathog. 2017;13:e1006514.
Zimmermann S, Pfannkuch L, Al-Zeer MA, Bartfeld S, Koch M, Liu J, Rechner C, Soerensen M, Sokolova O, Zamyatina A, et al. ALPK1- and TIFA-dependent innate immune response triggered by the helicobacter pylori type IV secretion system. Cell Rep. 2017;20:2384–95.
Crespo A, Filla MB, Russell SW, Murphy WJ. Indirect induction of suppressor of cytokine signalling-1 in macrophages stimulated with bacterial lipopolysaccharide: partial role of autocrine/paracrine interferon-alpha/beta. Biochem J. 2000;349:99–104.
Dalpke AH, Opper S, Zimmermann S, Heeg K. Suppressors of cytokine signaling (SOCS)-1 and SOCS-3 are induced by CpG-DNA and modulate cytokine responses in APCs. J Immunol. 2001;166:7082–9.
Yang T, Stark P, Janik K, Wigzell H, Rottenberg ME. SOCS-1 protects against chlamydia pneumoniae-induced lethal inflammation but hampers effective bacterial clearance. J Immunol. 2008;180:4040–9.
Ekchariyawat P, Pudla S, Limposuwan K, Arjcharoen S, Sirisinha S, Utaisincharoen P. Burkholderia pseudomallei-induced expression of suppressor of cytokine signaling 3 and cytokine-inducible src homology 2-containing protein in mouse macrophages: a possible mechanism for suppression of the response to gamma interferon stimulation. Infect Immun. 2005;73:7332–9.
Carow B, Ye X, Gavier-Widen D, Bhuju S, Oehlmann W, Singh M, Skold M, Ignatowicz L, Yoshimura A, Wigzell H, Rottenberg ME. Silencing suppressor of cytokine signaling-1 (SOCS1) in macrophages improves mycobacterium tuberculosis control in an interferon-gamma (IFN-gamma)-dependent manner. J Biol Chem. 2011;286:26873–87.
Wang X, Yang L, Huang F, Zhang Q, Liu S, Ma L, You Z. Inflammatory cytokines IL-17 and TNF-α up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol Lett. 2017;184:7–14.
Duffield AS, Ascierto ML, Anders RA, Taube JM, Meeker AK, Chen S, McMiller TL, Phillips NA, Xu H, Ogurtsova A, et al. Th17 immune microenvironment in Epstein-Barr virus-negative Hodgkin lymphoma: implications for immunotherapy. Blood advances. 2017;1:1324–34.
Zhang Y, Ma CA, Lawrence MG, Break TJ, O'Connell MP, Lyons JJ, Lopez DB, Barber JS, Zhao Y, Barber DL, et al. PD-L1 up-regulation restrains Th17 cell differentiation in STAT3 loss- and STAT1 gain-of-function patients. J Exp Med. 2017;214:2523–33.
Gao Y, Basile JI, Classon C, Gavier-Widen D, Yoshimura A, Carow B, Rottenberg ME. STAT3 expression by myeloid cells is detrimental for the T- cell-mediated control of infection with mycobacterium tuberculosis. PLoS Pathog. 2018;14:e1006809.
Funding
This work was supported by the Austrian Science Fund (FWF, Grant P 29941), the American Association of Immunologists, Grant AI0615_P and the Cancer Cluster Salzburg (CCS) Grant Nr. 20102-P1601064-FPR01–2017.
Author information
Authors and Affiliations
Contributions
TN, MS and JHH conceived and designed the experiments and wrote the paper, MS and TN performed the experiments, JV and SS analyzed the public genomic datasets, MMK IKG analyzed data and contributed to co-culture experiments, GP and SW analyzed data and contributed to the H. pylori infection experiments, JHH obtained funding for the study. All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All studies involving human cells were conducted in accordance with the guidelines of the World Medical Association’s Declaration of Helsinki. Since national regulations do not require informed consent in the case of anonymous blood cells discarded after plasmapheresis (buffy coats), no additional approval by the local ethics committee was required.
Consent for publication
Not applicable.
Competing interests
None of the authors report any conflicts of interest related to this work.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1 Figure S1
. H. pylori induces maturation of human DCs. Monocytes were isolated from human PBMCs and differentiated into moDCs in the presence of IL-4 and GMCSF (50 ng/mL each). Immature DCs were re-plated in fresh medium on day 7,H. pylori strain P12 was harvested in PBS and added to the cells at increasing MOIs of 0.2, 2 and 20. Surface marker expression (A) and cytokine secretion(B) were analyzed 48 h post-infection. Data represent mean + SD of one experiment comprising three individual donors (N = 3). For statistical analysis, one-way ANOVA with Dunnett’s post-hoc test was performed. *p < 0.05, **p < 0.01, ***p < 0.001. Figure S2.H. pylori-inducedSOCS3 expression is independent of MEK signaling. Cells were treated with a specific inhibitor of MEK kinase 20 min prior to infection with H. pylori wild-type strain P12 (MOI = 5). After 1 h of infection, DCs were harvested and SOCS3 mRNA expression was analyzed by qRT-PCR. Two experiments comprising 4 donors (N = 4) are shown. For statistical analysis, one-way ANOVA with Tukey’s post-hoc test was performed. *p < 0.05, **p < 0.01. Figure S3. SOCS3 silencing does not affect chemokine secretion. Immature day-7 DCs were re-plated and transfected with siRNA targeting SOCS3 or non-targeting control oligo (both 100 pmol). 48 h post-transfection, H. pylori strain P12 was harvested in PBS and added to the cells at MOI = 5. Surface marker expression(A) and chemokine secretion (B) were analyzed 24 h after bacterial infection. Three experiments comprising eight donors are shown. Dots represent individual donors; mean ± SD is shown. Figure S4.SOCS1 and SOCS2 silencing do not significantly alter DC activation. Immature day-7 DCs were re-plated and transfected with siRNA targeting SOCS1 (50–100 pmol), SOCS2 (100 pmol) or non-targeting control oligo (100 pmol). 48 h post-transfection, H. pylori strain P12 was harvested in PBS and added to the cells at MOI = 5. (A,C) Silencing efficiency was analyzed by qRT-PCR after 8 h of infection. (B,D) Surface marker expression was analyzed 24 h after bacterial infection. Three experiments comprising four donors (N = 4) (A,B) and three experiments comprising five donors (N = 5) (C,D) are shown. Dots represent individual donors; mean ± SD is shown. For statistical analysis, one-way ANOVA with Tukey’s post-hoc test was performed. *p < 0.05, **p < 0.01.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sarajlic, M., Neuper, T., Vetter, J. et al. H. pylori modulates DC functions via T4SS/TNFα/p38-dependent SOCS3 expression. Cell Commun Signal 18, 160 (2020). https://doi.org/10.1186/s12964-020-00655-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-020-00655-1